


よむ、つかう、まなぶ。
資料1-1 要指導医薬品のリスク評価について[205KB] (2 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_44431.html |
| 出典情報 | 薬事審議会 医薬品等安全対策部会安全対策調査会(令和6年度第8回 10/29)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
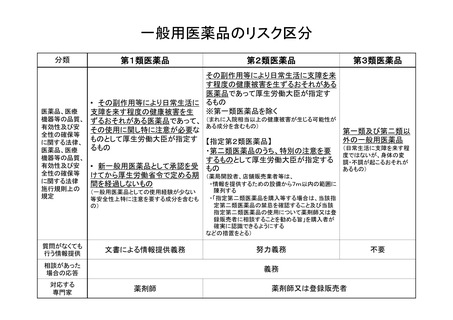
スイッチOTC薬等のリスク評価について
平成25年12月20日
医薬品等安全対策部会
1. 本年 12 月 13 日に公布された薬事法及び薬剤師法の一部を改正する法律(平成 25
年法律第 103 号)による改正後の薬事法(昭和 35 年法律第 145 号)第4条第5項第4
号に規定する要指導医薬品のうち、スイッチOTC薬及びダイレクトOTC薬につい
ては、一定の期間が経過することにより一般用医薬品に移行することとなるため、移
行の際には、一般用医薬品としての販売可否を確認するためのリスク評価を行う必要
がある。
2.
この評価については、原則3年間の製造販売後調査の終了までに行うこととし、
製造販売後2年以降の時点において、評価対象の医薬品の製造販売後調査の中間報告
の結果及び薬事法第 77 条の4の2に基づき報告される副作用報告を基に、重篤な副作
用の発生状況を評価し、製造販売承認の拒否事由に該当する状況にないことを確認し、
その後、製造販売後調査が終了するまでの間、当該評価が変わらないことを厚生労働
省に確認させることにより、製造販売後調査の終了時点で、要指導医薬品から一般用
医薬品へ移行するものとする。
3.
また、医薬品等安全対策部会は、一般用医薬品の区分の指定及びその変更に関す
る事項その他医薬品の安全性の確保に関する事項を調査審議するとされており、上記
の一般用医薬品としての販売可否の確認についても所掌することとなるが、この確認
手続については、今後、医薬品等安全対策部会長の了解を得て、安全対策調査会に行
わせることとし、その結果を医薬品等安全対策部会に報告させることとする。
4.
なお、リスク区分を決定するためのリスク評価は、上記の確認手続と異なり、死
亡や重篤以外の副作用やその発生頻度も含めて評価した上で、他の医薬品との比較を
しつつ検討するなど、より詳細な評価を行う必要があることから、従来と同様の取扱
いとする。
5.
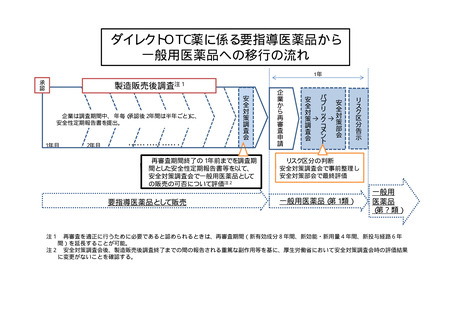
ダイレクトOTC薬については、スイッチOTC薬と異なり、新規に開発された
医療用医薬品と同様に使用経験がなく、副作用発生頻度の年次毎の変動や長期服薬時
の安全性等を確認する必要があること等を踏まえ、これまでと同様に4〜8年の再審
査期間で製造販売後調査を行うこととし、評価手続の期間を短縮することにより、再
審査期間終了時点で一般用医薬品としての販売可否の評価を行い、問題が無いことが
確認されれば、要指導医薬品から一般用医薬品へ移行するものとする。
以上
2 / 5
平成25年12月20日
医薬品等安全対策部会
1. 本年 12 月 13 日に公布された薬事法及び薬剤師法の一部を改正する法律(平成 25
年法律第 103 号)による改正後の薬事法(昭和 35 年法律第 145 号)第4条第5項第4
号に規定する要指導医薬品のうち、スイッチOTC薬及びダイレクトOTC薬につい
ては、一定の期間が経過することにより一般用医薬品に移行することとなるため、移
行の際には、一般用医薬品としての販売可否を確認するためのリスク評価を行う必要
がある。
2.
この評価については、原則3年間の製造販売後調査の終了までに行うこととし、
製造販売後2年以降の時点において、評価対象の医薬品の製造販売後調査の中間報告
の結果及び薬事法第 77 条の4の2に基づき報告される副作用報告を基に、重篤な副作
用の発生状況を評価し、製造販売承認の拒否事由に該当する状況にないことを確認し、
その後、製造販売後調査が終了するまでの間、当該評価が変わらないことを厚生労働
省に確認させることにより、製造販売後調査の終了時点で、要指導医薬品から一般用
医薬品へ移行するものとする。
3.
また、医薬品等安全対策部会は、一般用医薬品の区分の指定及びその変更に関す
る事項その他医薬品の安全性の確保に関する事項を調査審議するとされており、上記
の一般用医薬品としての販売可否の確認についても所掌することとなるが、この確認
手続については、今後、医薬品等安全対策部会長の了解を得て、安全対策調査会に行
わせることとし、その結果を医薬品等安全対策部会に報告させることとする。
4.
なお、リスク区分を決定するためのリスク評価は、上記の確認手続と異なり、死
亡や重篤以外の副作用やその発生頻度も含めて評価した上で、他の医薬品との比較を
しつつ検討するなど、より詳細な評価を行う必要があることから、従来と同様の取扱
いとする。
5.
ダイレクトOTC薬については、スイッチOTC薬と異なり、新規に開発された
医療用医薬品と同様に使用経験がなく、副作用発生頻度の年次毎の変動や長期服薬時
の安全性等を確認する必要があること等を踏まえ、これまでと同様に4〜8年の再審
査期間で製造販売後調査を行うこととし、評価手続の期間を短縮することにより、再
審査期間終了時点で一般用医薬品としての販売可否の評価を行い、問題が無いことが
確認されれば、要指導医薬品から一般用医薬品へ移行するものとする。
以上
2 / 5