



よむ、つかう、まなぶ。
参考資料6 新型コロナワクチン「スパイクバックス筋注(1価:起源株)」(モデルナ・ジャパン株式会社)添付文書 (3 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/shingi2/0000208910_00056.html |
| 出典情報 | 第89回厚生科学審議会予防接種・ワクチン分科会副反応検討部会、令和4年度第21回薬事・食品衛生審議会薬事分科会医薬品等安全対策部会安全対策調査会(合同開催)(12/16)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
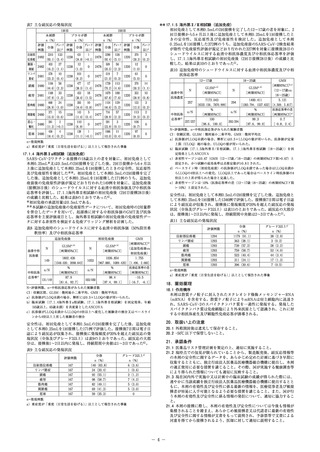
集積した時点で実施し、SARS‑CoV‑2による感染症に対するVEを評価し
た。解析結果は表1のとおりであった。なお、中間解析時及び主要解析時
の2回目接種後の追跡期間(中央値)はそれぞれ49日と64日であった3)。
安全性は、治験薬を少なくとも1回接種した200例で評価した。接種後7日
間は電子日誌により副反応が収集され、本剤群でいずれかの接種後に発現
頻度が20%を超えた副反応の発現状況(全体及びグレード3以上)は表4の
とおりであった。副反応の大部分は、接種後1〜2日以内に発現し、持続期
間中央値は1〜3日であった4)。
表1 SARS‑CoV‑2による感染症に対する有効性
本剤群
COVID‑19
確定例
(例)
13934
5
14134
11
解析対象
(例)
中間解析
主要解析
プラセボ群
COVID‑19
VE(%)
確定例
[信頼区間]a)
(例)
13883
90
94.5[81.8, 98.3]
14073
185
94.1[89.3, 96.8]
表4 主な副反応の発現状況
解析対象
(例)
1回目
2回目
本剤群(N=150) プラセボ群(N=50) 本剤群(N=147) プラセボ群(N=50)
n(%)
n(%)
n(%)
n(%)
グレード
グレード
グレード
グレード
全体
全体
全体
全体
3以上a)
3以上a)
3以上a)
3以上a)
注射部位
124
2
3
125
6
1
0
0
疼痛 (82.7) (1.3) (6.0)
(85.0) (4.1) (2.0)
20
70
10
5
頭痛
0
0
0
0
(13.3)
(47.6) (6.8) (10.0)
28
5
93
26
4
疲労
0
0
0
(18.7)
(10.0)
(63.3) (17.7) (8.0)
56
1
2
73
10
5
筋肉痛
0
0
(37.3) (0.7) (4.0)
(49.7) (6.8) (10.0)
12
47
11
関節痛
0
0
0
0
0
(8.0)
(32.0) (7.5)
8
1
74
7
悪寒
0
0
0
0
(5.3)
(2.0)
(50.3) (4.8)
3
1
1
1
59
8
発熱b)
0
0
(2.0) (0.7) (2.0) (2.0) (40.1) (5.4)
COVID‑19確定例:RT‑PCR検査陽性かつ2つ以上の全身症状又は1つ以上の呼吸器症
状を呈する症候性COVID‑19で、2回目接種から14日後以降に発症した症例
VEの解析には接種間隔21〜42日間の被験者が含まれ、そのうち接種間隔が25〜35日間
の被験者が中間解析では93.0%(25861例)
、主要解析では97.7%(27567例)であった。
a)投与群を共変量とし、年齢とCOVID‑19重症化リスク(18〜64歳かつ重症化リスク
因子なし、18〜64歳かつ重症化リスク因子あり、65歳以上)を層別因子とした層
別Cox比例ハザードモデルにより算出。中間解析は99.1%信頼区間、主要解析
は95%信頼区間。
安全性は、治験薬を少なくとも1回接種した30351例で評価した。各接種後
7日間は電子日誌により副反応が収集され、本剤群でいずれかの接種後に
発現頻度が20%を超えた又はグレード4が複数例に発現した副反応の発現
状況(全体及びグレード3以上)は表2のとおりであった。副反応の大部分
は、接種後1〜2日以内に発現し、持続期間中央値は1〜3日であった3)。
表2 主な副反応の発現状況
注射部
位疼痛
頭痛
疲労
筋肉痛
関節痛
悪寒
発熱b)
1回目
2回目
本剤群
プラセボ群
本剤群
プラセボ群
n(%)
n(%)
n(%)
n(%)
評価
グレード 評価
グレード 評価
グレード 評価
グレード
全体
全体
全体
全体
例数
3以上a) 例数
3以上a) 例数
3以上a) 例数
3以上a)
12690 416
2658
55
12943 604
2477
40
15164
15151
14673
14562
(83.7)(2.7)
(17.5)(0.4)
(88.2)(4.1)
(17.0)(0.3)
4951 271
4027 196
8602 659
3410 162
15163
15150
14673
14562
(32.7)(1.8)
(26.6)(1.3)
(58.6)(4.5)
(23.4)(1.1)
5635 151
4133 105
9582 1428
3403 106
15163
15150
14673
14560
(37.2)(1.0)
(27.3)(0.7)
(65.3)(9.7)
(23.4)(0.7)
3441
90
2071
47
8508 1318
1809
52
15163
15150
14673
14560
(22.7)(0.6)
(13.7)(0.3)
(58.0)(9.0)
(12.4)(0.4)
2511
61
1783
37
6284 770
1569
44
15163
15150
14673
14560
(16.6)(0.4)
(11.8)(0.2)
(42.8)(5.2)
(10.8)(0.3)
1253
24
878
14
6482 191
809
17
15163
15150
14673
14560
(8.3)(0.2)
(5.8)(<0.1)
(44.2)(1.3)
(5.6)(0.1)
115
15
44
8
2278 215
43
5
15164
15153
14669
14559
(0.8)(0.1)
(0.3)(<0.1)
(15.5)(1.5)
(0.3)(<0.1)
N=評価例数(電子日誌により評価した例数)、n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
b)口腔内体温が38℃以上。39℃以上を重症度が重度(グレード3)以上とした。
17.1.3 海外第Ⅱ/Ⅲ相試験(初回免疫)
SARS‑CoV‑2ワクチン未接種の12〜17歳の者を対象に、無作為化プラセボ対
照観察者盲検の第Ⅱ/Ⅲ相試験を実施し、本剤又はプラセボ0.5mLを4週間隔
で2回筋肉内接種したときの安全性、免疫原性及び有効性を検討した。本試
験には本剤群2489例及びプラセボ群1243例が組み入れられた。主要評価項目
である免疫原性は、ベースライン時のSARS‑CoV‑2感染が否定され、規定さ
れた2回目接種を受けた本剤群の340例を対象に評価し、17.1.1海外第Ⅲ相試
験の本剤群のうち18〜25歳の被験者データと比較した。本剤2回目接種から
28日後のシュードウイルスに対する血清中和抗体価及び中和抗体応答率は表
5のとおりであり、12〜17歳の18〜25歳に対する非劣性が確認された5)。
n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
b)口腔内体温が38℃以上。39℃以上をグレード3以上とした。
17.1.2 国内第Ⅰ/Ⅱ相試験(初回免疫)
SARS‑CoV‑2ワクチン未接種の20歳以上の日本人健康成人を対象に、無作為
化プラセボ対照観察者盲検の第Ⅰ/Ⅱ相臨床試験を実施し、本剤又はプラセ
ボ0.5mLを4週間隔で2回筋肉内接種したときの安全性及び免疫原性を検討し
た。本試験には本剤群150例及びプラセボ群50例が組み入れられ、2回目接種
から28日後のSARS‑CoV‑2に対する血清結合抗体価及び野生型ウイルスに対
する血清中和抗体価の幾何平均値(GMT)
、幾何平均増加倍率(GMFR)及
び抗体陽転率(SCR)が検討された。結果は表3のとおりであった4)。
表3 2回目接種28日後のSARS‑CoV‑2血清結合抗体価及び血清中和抗体価
血清結合抗体価
全年齢
本剤群
20〜64歳
65歳以上
プラセボ群
全年齢
血清中和抗体価
全年齢
本剤群
20〜64歳
65歳以上
プラセボ群
全年齢
SCR
%
n
[両側95%CI]a)
100
813.05
1009.25
147
147
[97.5, 100.0]
[759.31, 870.60][865.11, 1177.40]
810.61
1037.79
100
98
98
[750.45, 875.60][867.37, 1241.69]
[96.3, 100.0]
817.95
954.51
100
49
49
[711.35, 940.52][706.61, 1289.37]
[92.7, 100.0]
0.60
0.90
2.0
49
1
[0.53, 0.68]
[0.83, 0.98]
[0.1, 10.9]
SCR
GMT
GMFR
%
N
n
[両側95%CI]
[両側95%CI]
[両側95%CI]a)
1731.1
21.7
100
146
146
[1579.0, 1897.8] [19.8, 23.8]
[97.5, 100.0]
1727.4
21.6
100
97
97
[1549.0, 1926.5] [19.4, 24.1]
[96.3, 100.0]
1738.3
21.8
100
49
49
[1459.9, 2069.8] [18.3, 25.9]
[92.7, 100.0]
79.9
1.0
0
49
0
[79.9, 79.9]
[1.0, 1.0]
[0.0, 7.3]
N
GMT
[両側95%CI]
GMFR
[両側95%CI]
N=評価例数、n=抗体陽転例数
CI:信頼区間、GMT:幾何平均値、GMFR:幾何平均増加倍率、SCR:抗体陽転率
a)抗体価が検出限界(LOD)又は定量下限(LLOQ)未満からLOD又はLLOQ以上へ
変化した被験者の割合、又は、ベースラインから4倍以上上昇した被験者の割合
−3−
表5 2回目接種28日後のシュードウイルスに対する血清中和抗体価(50%
阻害希釈倍率)及び中和抗体応答率
年齢
12〜17歳
18〜25歳
GMR
[両側95%CI]b,c)
N
N
(12〜17歳vs
血清中和
18〜25歳)
抗体価
1401.670
1301.312
1.077
340
296
[1276.300, 1539.355]
[1176.979, 1438.780][0.939, 1.236]
%
%
抗体応答率の差
n/N
n/N
e)
中和抗体
[両側95%CI]
[両側95%CI] [両側95%CI]
98.8
98.6
0.2
応答率d)
336/340
292/296
[97.0, 99.7]
[96.6, 99.6]
[‑1.8, 2.4]
GLSMa,b)
[両側95%CI]
GLSMa,b)
[両側95%CI]
N=評価例数、n=中和抗体応答がみられた被験者数
CI:信頼区間、GLSM:幾何最小二乗平均、GMR:幾何平均比
a)抗体価がLLOQ未満の場合、解析には0.5×LLOQの値が用いられた。
b)臨床試験(17.1.3海外第Ⅱ/Ⅲ相試験、17.1.1海外第Ⅲ相試験(18〜25歳))を固
定効果としたANCOVA
c)非劣性マージンは0.67(GMR(12〜17歳/18〜25歳)の両側95%CI下限>0.67)と
設定され、かつ試験の成功基準は点推定値が>0.8とされた。
d)抗体価がLLOQ未満からLLOQ以上へ変化した被験者の割合又はベースラインから
3.3倍以上上昇した被験者の割合
e)非劣性マージンは‑10%(抗体応答率の差(12〜17歳‑18〜25歳)の両側95%CI下限
>‑10%)と設定され、かつ試験の成功基準は点推定値が>‑5%とされた。
副次評価項目であるワクチンの有効性(VE)は、ベースライン時の
SARS‑CoV‑2感染が否定され、2回目接種後14日以降に発症した
COVID‑19確定例を対象に評価した。データカットオフ日時点のSARS‑
CoV‑2による感染症に対するVEは表6のとおりであった。データカットオ
フ日時点で、2回目接種後の追跡期間(中央値)は53日であった5)。
表6 SARS‑CoV‑2による感染症に対する有効性
本剤群
COVID‑19
解析対象
確定例
(例)
(例)
2139
0
プラセボ群
COVID‑19
解析対象
確定例
(例)
(例)
1042
4
VE(%)
[両側95%CI]
100
[28.9, NE]
NE:評価不能、CI:信頼区間
COVID‑19確定例:RT‑PCR検査陽性かつ2つ以上の全身症状又は1つ以上の呼吸器症
状を呈する症候性COVID‑19で、2回目接種から14日後以降に発症した症例
安全性は、治験薬を少なくとも1回接種した3726例で評価した。各接種後7
日間は電子日誌により副反応が収集され、本剤群でいずれかの接種後に発
現頻度が20%を超えた又はグレード4が複数例に発現した副反応の発現状
況(全体及びグレード3以上)は表7のとおりであった。副反応の大部分
は、接種後1〜2日以内に発現し、持続期間中央値は1〜3日であった5)。
た。解析結果は表1のとおりであった。なお、中間解析時及び主要解析時
の2回目接種後の追跡期間(中央値)はそれぞれ49日と64日であった3)。
安全性は、治験薬を少なくとも1回接種した200例で評価した。接種後7日
間は電子日誌により副反応が収集され、本剤群でいずれかの接種後に発現
頻度が20%を超えた副反応の発現状況(全体及びグレード3以上)は表4の
とおりであった。副反応の大部分は、接種後1〜2日以内に発現し、持続期
間中央値は1〜3日であった4)。
表1 SARS‑CoV‑2による感染症に対する有効性
本剤群
COVID‑19
確定例
(例)
13934
5
14134
11
解析対象
(例)
中間解析
主要解析
プラセボ群
COVID‑19
VE(%)
確定例
[信頼区間]a)
(例)
13883
90
94.5[81.8, 98.3]
14073
185
94.1[89.3, 96.8]
表4 主な副反応の発現状況
解析対象
(例)
1回目
2回目
本剤群(N=150) プラセボ群(N=50) 本剤群(N=147) プラセボ群(N=50)
n(%)
n(%)
n(%)
n(%)
グレード
グレード
グレード
グレード
全体
全体
全体
全体
3以上a)
3以上a)
3以上a)
3以上a)
注射部位
124
2
3
125
6
1
0
0
疼痛 (82.7) (1.3) (6.0)
(85.0) (4.1) (2.0)
20
70
10
5
頭痛
0
0
0
0
(13.3)
(47.6) (6.8) (10.0)
28
5
93
26
4
疲労
0
0
0
(18.7)
(10.0)
(63.3) (17.7) (8.0)
56
1
2
73
10
5
筋肉痛
0
0
(37.3) (0.7) (4.0)
(49.7) (6.8) (10.0)
12
47
11
関節痛
0
0
0
0
0
(8.0)
(32.0) (7.5)
8
1
74
7
悪寒
0
0
0
0
(5.3)
(2.0)
(50.3) (4.8)
3
1
1
1
59
8
発熱b)
0
0
(2.0) (0.7) (2.0) (2.0) (40.1) (5.4)
COVID‑19確定例:RT‑PCR検査陽性かつ2つ以上の全身症状又は1つ以上の呼吸器症
状を呈する症候性COVID‑19で、2回目接種から14日後以降に発症した症例
VEの解析には接種間隔21〜42日間の被験者が含まれ、そのうち接種間隔が25〜35日間
の被験者が中間解析では93.0%(25861例)
、主要解析では97.7%(27567例)であった。
a)投与群を共変量とし、年齢とCOVID‑19重症化リスク(18〜64歳かつ重症化リスク
因子なし、18〜64歳かつ重症化リスク因子あり、65歳以上)を層別因子とした層
別Cox比例ハザードモデルにより算出。中間解析は99.1%信頼区間、主要解析
は95%信頼区間。
安全性は、治験薬を少なくとも1回接種した30351例で評価した。各接種後
7日間は電子日誌により副反応が収集され、本剤群でいずれかの接種後に
発現頻度が20%を超えた又はグレード4が複数例に発現した副反応の発現
状況(全体及びグレード3以上)は表2のとおりであった。副反応の大部分
は、接種後1〜2日以内に発現し、持続期間中央値は1〜3日であった3)。
表2 主な副反応の発現状況
注射部
位疼痛
頭痛
疲労
筋肉痛
関節痛
悪寒
発熱b)
1回目
2回目
本剤群
プラセボ群
本剤群
プラセボ群
n(%)
n(%)
n(%)
n(%)
評価
グレード 評価
グレード 評価
グレード 評価
グレード
全体
全体
全体
全体
例数
3以上a) 例数
3以上a) 例数
3以上a) 例数
3以上a)
12690 416
2658
55
12943 604
2477
40
15164
15151
14673
14562
(83.7)(2.7)
(17.5)(0.4)
(88.2)(4.1)
(17.0)(0.3)
4951 271
4027 196
8602 659
3410 162
15163
15150
14673
14562
(32.7)(1.8)
(26.6)(1.3)
(58.6)(4.5)
(23.4)(1.1)
5635 151
4133 105
9582 1428
3403 106
15163
15150
14673
14560
(37.2)(1.0)
(27.3)(0.7)
(65.3)(9.7)
(23.4)(0.7)
3441
90
2071
47
8508 1318
1809
52
15163
15150
14673
14560
(22.7)(0.6)
(13.7)(0.3)
(58.0)(9.0)
(12.4)(0.4)
2511
61
1783
37
6284 770
1569
44
15163
15150
14673
14560
(16.6)(0.4)
(11.8)(0.2)
(42.8)(5.2)
(10.8)(0.3)
1253
24
878
14
6482 191
809
17
15163
15150
14673
14560
(8.3)(0.2)
(5.8)(<0.1)
(44.2)(1.3)
(5.6)(0.1)
115
15
44
8
2278 215
43
5
15164
15153
14669
14559
(0.8)(0.1)
(0.3)(<0.1)
(15.5)(1.5)
(0.3)(<0.1)
N=評価例数(電子日誌により評価した例数)、n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
b)口腔内体温が38℃以上。39℃以上を重症度が重度(グレード3)以上とした。
17.1.3 海外第Ⅱ/Ⅲ相試験(初回免疫)
SARS‑CoV‑2ワクチン未接種の12〜17歳の者を対象に、無作為化プラセボ対
照観察者盲検の第Ⅱ/Ⅲ相試験を実施し、本剤又はプラセボ0.5mLを4週間隔
で2回筋肉内接種したときの安全性、免疫原性及び有効性を検討した。本試
験には本剤群2489例及びプラセボ群1243例が組み入れられた。主要評価項目
である免疫原性は、ベースライン時のSARS‑CoV‑2感染が否定され、規定さ
れた2回目接種を受けた本剤群の340例を対象に評価し、17.1.1海外第Ⅲ相試
験の本剤群のうち18〜25歳の被験者データと比較した。本剤2回目接種から
28日後のシュードウイルスに対する血清中和抗体価及び中和抗体応答率は表
5のとおりであり、12〜17歳の18〜25歳に対する非劣性が確認された5)。
n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
b)口腔内体温が38℃以上。39℃以上をグレード3以上とした。
17.1.2 国内第Ⅰ/Ⅱ相試験(初回免疫)
SARS‑CoV‑2ワクチン未接種の20歳以上の日本人健康成人を対象に、無作為
化プラセボ対照観察者盲検の第Ⅰ/Ⅱ相臨床試験を実施し、本剤又はプラセ
ボ0.5mLを4週間隔で2回筋肉内接種したときの安全性及び免疫原性を検討し
た。本試験には本剤群150例及びプラセボ群50例が組み入れられ、2回目接種
から28日後のSARS‑CoV‑2に対する血清結合抗体価及び野生型ウイルスに対
する血清中和抗体価の幾何平均値(GMT)
、幾何平均増加倍率(GMFR)及
び抗体陽転率(SCR)が検討された。結果は表3のとおりであった4)。
表3 2回目接種28日後のSARS‑CoV‑2血清結合抗体価及び血清中和抗体価
血清結合抗体価
全年齢
本剤群
20〜64歳
65歳以上
プラセボ群
全年齢
血清中和抗体価
全年齢
本剤群
20〜64歳
65歳以上
プラセボ群
全年齢
SCR
%
n
[両側95%CI]a)
100
813.05
1009.25
147
147
[97.5, 100.0]
[759.31, 870.60][865.11, 1177.40]
810.61
1037.79
100
98
98
[750.45, 875.60][867.37, 1241.69]
[96.3, 100.0]
817.95
954.51
100
49
49
[711.35, 940.52][706.61, 1289.37]
[92.7, 100.0]
0.60
0.90
2.0
49
1
[0.53, 0.68]
[0.83, 0.98]
[0.1, 10.9]
SCR
GMT
GMFR
%
N
n
[両側95%CI]
[両側95%CI]
[両側95%CI]a)
1731.1
21.7
100
146
146
[1579.0, 1897.8] [19.8, 23.8]
[97.5, 100.0]
1727.4
21.6
100
97
97
[1549.0, 1926.5] [19.4, 24.1]
[96.3, 100.0]
1738.3
21.8
100
49
49
[1459.9, 2069.8] [18.3, 25.9]
[92.7, 100.0]
79.9
1.0
0
49
0
[79.9, 79.9]
[1.0, 1.0]
[0.0, 7.3]
N
GMT
[両側95%CI]
GMFR
[両側95%CI]
N=評価例数、n=抗体陽転例数
CI:信頼区間、GMT:幾何平均値、GMFR:幾何平均増加倍率、SCR:抗体陽転率
a)抗体価が検出限界(LOD)又は定量下限(LLOQ)未満からLOD又はLLOQ以上へ
変化した被験者の割合、又は、ベースラインから4倍以上上昇した被験者の割合
−3−
表5 2回目接種28日後のシュードウイルスに対する血清中和抗体価(50%
阻害希釈倍率)及び中和抗体応答率
年齢
12〜17歳
18〜25歳
GMR
[両側95%CI]b,c)
N
N
(12〜17歳vs
血清中和
18〜25歳)
抗体価
1401.670
1301.312
1.077
340
296
[1276.300, 1539.355]
[1176.979, 1438.780][0.939, 1.236]
%
%
抗体応答率の差
n/N
n/N
e)
中和抗体
[両側95%CI]
[両側95%CI] [両側95%CI]
98.8
98.6
0.2
応答率d)
336/340
292/296
[97.0, 99.7]
[96.6, 99.6]
[‑1.8, 2.4]
GLSMa,b)
[両側95%CI]
GLSMa,b)
[両側95%CI]
N=評価例数、n=中和抗体応答がみられた被験者数
CI:信頼区間、GLSM:幾何最小二乗平均、GMR:幾何平均比
a)抗体価がLLOQ未満の場合、解析には0.5×LLOQの値が用いられた。
b)臨床試験(17.1.3海外第Ⅱ/Ⅲ相試験、17.1.1海外第Ⅲ相試験(18〜25歳))を固
定効果としたANCOVA
c)非劣性マージンは0.67(GMR(12〜17歳/18〜25歳)の両側95%CI下限>0.67)と
設定され、かつ試験の成功基準は点推定値が>0.8とされた。
d)抗体価がLLOQ未満からLLOQ以上へ変化した被験者の割合又はベースラインから
3.3倍以上上昇した被験者の割合
e)非劣性マージンは‑10%(抗体応答率の差(12〜17歳‑18〜25歳)の両側95%CI下限
>‑10%)と設定され、かつ試験の成功基準は点推定値が>‑5%とされた。
副次評価項目であるワクチンの有効性(VE)は、ベースライン時の
SARS‑CoV‑2感染が否定され、2回目接種後14日以降に発症した
COVID‑19確定例を対象に評価した。データカットオフ日時点のSARS‑
CoV‑2による感染症に対するVEは表6のとおりであった。データカットオ
フ日時点で、2回目接種後の追跡期間(中央値)は53日であった5)。
表6 SARS‑CoV‑2による感染症に対する有効性
本剤群
COVID‑19
解析対象
確定例
(例)
(例)
2139
0
プラセボ群
COVID‑19
解析対象
確定例
(例)
(例)
1042
4
VE(%)
[両側95%CI]
100
[28.9, NE]
NE:評価不能、CI:信頼区間
COVID‑19確定例:RT‑PCR検査陽性かつ2つ以上の全身症状又は1つ以上の呼吸器症
状を呈する症候性COVID‑19で、2回目接種から14日後以降に発症した症例
安全性は、治験薬を少なくとも1回接種した3726例で評価した。各接種後7
日間は電子日誌により副反応が収集され、本剤群でいずれかの接種後に発
現頻度が20%を超えた又はグレード4が複数例に発現した副反応の発現状
況(全体及びグレード3以上)は表7のとおりであった。副反応の大部分
は、接種後1〜2日以内に発現し、持続期間中央値は1〜3日であった5)。