



よむ、つかう、まなぶ。
参考資料6 新型コロナワクチン「スパイクバックス筋注(1価:起源株)」(モデルナ・ジャパン株式会社)添付文書 (4 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/shingi2/0000208910_00056.html |
| 出典情報 | 第89回厚生科学審議会予防接種・ワクチン分科会副反応検討部会、令和4年度第21回薬事・食品衛生審議会薬事分科会医薬品等安全対策部会安全対策調査会(合同開催)(12/16)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
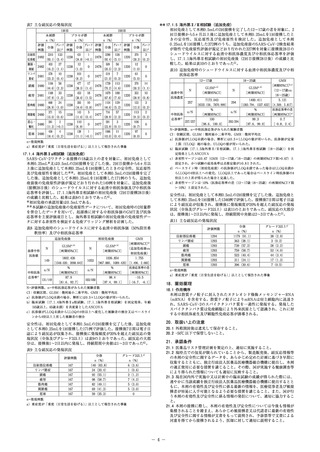
**17.1.5 海外第Ⅱ/Ⅲ相試験(追加免疫)
表7 主な副反応の発現状況
1回目
評価
例数
注射部
2482
位疼痛
腫脹・
2482
硬結
リンパ
2481
節症
頭痛 2480
疲労 2481
筋肉痛 2480
関節痛 2480
悪心・
2480
嘔吐
悪寒 2480
初回免疫として本剤0.5mLの2回接種を完了した12〜17歳の者を対象に、2
回目接種から5ヵ月以上後に追加免疫として本剤0.25mLを1回接種したと
きの安全性、反応原性及び免疫原性を検討した。追加免疫として本剤
0.25mLを1回接種した372例のうち、追加免疫前のSARS‑CoV‑2検査結果
が陰性で免疫原性評価が規定どおり行われた327例を対象に接種後28日の
シュードウイルスに対する血清中和抗体濃度及び中和抗体応答率を評価
し、17.1.1海外第Ⅲ相試験の初回免疫後(2回目接種28日後)の成績と比
較した。結果は表10のとおりであった5)。
2回目
本剤群
プラセボ群
n(%)
n(%)
グレード 評価
グレード 評価
全体
全体
3以上a) 例数
3以上a) 例数
2310 133
431
1
1238
2478
(93.1)(5.4)
(34.8)(<0.1)
403
27
12
1238
0
2478
(16.2)(1.1)
(1.0)
578
10
101
1238
0
2477
(23.3)(0.4)
(8.2)
1106
56
477
17
1238
2478
(44.6)(2.3)
(38.5)(1.4)
1188
33
453
18
1238
2478
(47.9)(1.3)
(36.6)(1.5)
668
24
205
10
1238
2477
(26.9)(1.0)
(16.6)(0.8)
371
15
143
5
1238
2477
(15.0)(0.6)
(11.6)(0.4)
281
2
110
1238
0
2477
(11.3)(<0.1)
(8.9)
456
4
138
1
1238
2477
(18.4)(0.2)
(11.1)(<0.1)
本剤群
プラセボ群
n(%)
n(%)
グレード 評価
グレード
全体
全体
3以上a) 例数
3以上a)
2290 126
370
3
1220
(92.4)(5.1)
(30.3)(0.2)
509
56
12
1220
0
(20.5)(2.3)
(1.0)
519
7
61
1220
0
(21.0)(0.3)
(5.0)
1739 113
370
14
1220
(70.2)(4.6)
(30.3)(1.1)
1679 188
353
10
1220
(67.8)(7.6)
(28.9)(0.8)
1154 129
153
3
1220
(46.6)(5.2)
(12.5)(0.2)
716
57
113
2
1220
(28.9)(2.3)
(9.3)(0.2)
591
3
106
1220
0
(23.9)(0.1)
(8.7)
1066
11
97
1220
0
(43.0)(0.4)
(8.0)
表10 追加免疫時のシュードウイルスに対する血清中和抗体濃度及び中和
抗体応答率
12〜17歳
GMR
[両側95%CI]b,c)
N
N
(12〜17歳vs
血清中和
18〜25歳)
抗体濃度
7172.043
1400.411
5.121
257
294
[6535.156, 7870.999]
[1283.794, 1527.622][4.509, 5.817]
%
%
抗体応答率の差
n/N
n/N
e)
中和抗体
[両側95%CI]
[両側95%CI] [両側95%CI]
100
99.3
0.7
応答率d)
257/257
292/294
[98.6, 100.0]
[97.6, 99.9]
[‑0.8, 2.4]
17.1.4 海外第Ⅱa相試験(追加免疫)
SARS‑CoV‑2ワクチン未接種の18歳以上の者を対象に、初回免疫として
本剤0.25mL※又は0.5mLの2回接種を完了した後、2回目接種から6ヵ月以
上後に追加免疫として本剤0.25mLを1回接種したときの安全性、反応原性
及び免疫原性を検討した※※。初回免疫として本剤0.5mLの2回接種を完了
した後、追加免疫として本剤0.25mLを1回接種した171例のうち、追加免
疫前後の免疫原性評価が規定どおり行われた149例を対象に、追加免疫後
(接種28日後)のシュードウイルスに対する血清中和抗体価及び中和抗体
応答率を評価し、17.1.1海外第Ⅲ相試験の初回免疫後(2回目接種28日後)
の成績と比較した。結果は表8のとおりであった6)。
※初回免疫の承認用量は0.5mLである。
※※本試験の追加免疫後の免疫原性データについて、初回免疫時の2用量群
を併合したデータを用いて、起源株に対する中和抗体価のGMT及び抗体
応答率を主要評価項目とし、海外第Ⅲ相試験の初回免疫後の免疫原性デー
タに対する非劣性を検証する免疫ブリッジング解析を計画した。
安全性は、初回免疫として本剤0.5mLの2回接種を完了した後、追加免疫と
して本剤0.25mLを1回接種した1346例で評価した。接種後7日間は電子日誌
により副反応が収集され、接種後に発現頻度が20%を超えた副反応の発現
状況(全体及びグレード3以上)は表11のとおりであった。副反応の大部分
は、接種後1〜2日以内に発現し、持続期間中央値は2〜3日であった5)。
表11 主な副反応の発現状況
表8 追加免疫時のシュードウイルスに対する血清中和抗体価(50%阻害希
釈倍率)及び中和抗体応答率
GMR
[両側95%CI]b)
GLSMa,b)
GLSMa,b)
N
N
(追加免疫後vs
血清中和
[両側95%CI]
[両側95%CI]
初回免疫後)
抗体価
1802.426
1026.854
1.755
149
1053
[1548.020, 2098.643]
[967.880, 1089.420][1.496, 2.060]
%
%
抗体応答率の差
n/N
n/N
中和抗体
[両側95%CI]
[両側95%CI]
[両側95%CI]
87.9
98.4
‑10.5
応答率c)
131/149
1033/1050
[81.6, 92.7]
[97.4, 99.1] [‑16.7, ‑6.1]
安全性は、初回免疫として本剤0.5mLの2回接種を完了した後、追加免疫
として本剤0.25mLを1回接種した171例で評価した。接種後7日間は電子日
誌により副反応が収集され、接種後に発現頻度が20%を超えた副反応の発
現状況(全体及びグレード3以上)は表9のとおりであった。副反応の大部
分は、接種後1〜2日以内に発現し、持続期間中央値は1〜3日であった6)。
表9 主な副反応の発現状況
評価例数
注射部位疼痛
リンパ節症
頭痛
疲労
筋肉痛
関節痛
悪寒
167
167
167
167
167
167
167
評価例数
注射部位疼痛
リンパ節症
頭痛
疲労
筋肉痛
関節痛
悪寒
初回免疫後
N=評価例数、n=中和抗体応答がみられた被験者数
CI:信頼区間、GLSM:幾何最小二乗平均、GMR:幾何平均比
a)抗体価がLLOQ未満の場合、解析には0.5×LLOQの値が用いられた。
b)臨床試験(17.1.4海外第Ⅱa相試験、17.1.1海外第Ⅲ相試験)を固定効果、年齢
(65歳以上、65歳未満)を共変量としたANCOVA
c)抗体価がLLOQ未満からLLOQの4倍以上へ変化した被験者の割合又はベースライ
ンから4倍以上上昇した被験者の割合
全体
n(%)
140(83.8)
34(20.4)
92(55.1)
98(58.7)
82(49.1)
69(41.3)
59(35.3)
グレード3以上a)
n(%)
6(3.6)
1(0.6)
2(1.2)
7(4.2)
5(3.0)
5(3.0)
0
n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
GLSMa,b)
[両側95%CI]
N=評価例数、n=中和抗体応答がみられた被験者数
CI:信頼区間、GLSM:幾何最小二乗平均、GMR:幾何平均比
a)抗体価がLLOQ未満の場合、解析には0.5×LLOQの値が用いられ、抗体価が定量
上限(ULOQ)超の場合、ULOQの値が用いられた。
b)臨床試験(17.1.5海外第Ⅱ/Ⅲ相試験、17.1.1海外第Ⅲ相試験(18〜25歳))を固
定効果としたANCOVA
c)非劣性マージンは0.67(GMR(12〜17歳/18〜25歳)の両側95%CI下限>0.67)と
設定され、かつ試験の成功基準は点推定値が≥0.8とされた。
d)ベースライン時(初回免疫前)の抗体価がLLOQ未満であった場合はLLOQ未満か
らLLOQの4倍以上への変化、LLOQ以上であった場合はベースライン時抗体価の4
倍以上の上昇が得られた場合と定義した。
e)非劣性マージンは‑10%(抗体応答率の差(12〜17歳‑18〜25歳)の両側95%CI下限
>‑10%)と設定された。
n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
追加免疫後
18〜25歳
GLSMa,b)
[両側95%CI]
1294
1293
1293
1293
1293
1293
1293
全体
n(%)
1179(91.1)
363(28.1)
739(57.2)
759(58.7)
523(40.4)
311(24.1)
396(30.6)
グレード3以上a)
n(%)
38(2.9)
3(0.2)
28(2.2)
52(4.0)
44(3.4)
17(1.3)
7(0.5)
n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
18. 薬効薬理
18.1 作用機序
本剤は脂質ナノ粒子に封入されたヌクレオシド修飾メッセンジャーRNA
(mRNA)を含有する。脂質ナノ粒子によりmRNAは宿主細胞内に送達さ
れ、SARS‑CoV‑2のスパイクタンパク質を一過性に発現する。発現した
スパイクタンパク質は免疫細胞により外来抗原として認識され、これに対
する中和抗体産生及び細胞性免疫応答が誘導される。
20. 取扱い上の注意
20.1 外箱開封後は遮光して保存すること。
20.2 ‑50℃以下で保管しないこと。
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 現時点での知見が限られていることから、製造販売後、副反応情報等
の本剤の安全性に関するデータを、あらかじめ定めた計画に基づき早期に
収集するとともに、独立行政法人医薬品医療機器総合機構に提出し、本剤
の適正使用に必要な措置を講じること。その際、国が実施する健康調査等
により得られた情報についても適切に反映すること。
21.3 現在国内外で実施中又は計画中の臨床試験の成績が得られた際には、
速やかに当該成績を独立行政法人医薬品医療機器総合機構に提出するとと
もに、本剤の有効性及び安全性に係る最新の情報を、医療従事者及び被接
種者が容易に入手可能となるよう必要な措置を講じること。また、国が行
う本剤の有効性及び安全性に係る情報の発信について、適切に協力するこ
と。
21.4 本剤の接種に際し、本剤の有効性及び安全性については今後も情報が
集積されることを踏まえ、あらかじめ被接種者又は代諾者に最新の有効性
及び安全性に関する情報が文書をもって説明され、予診票等で文書による
同意を得てから接種されるよう、医師に対して適切に説明すること。
−4−
表7 主な副反応の発現状況
1回目
評価
例数
注射部
2482
位疼痛
腫脹・
2482
硬結
リンパ
2481
節症
頭痛 2480
疲労 2481
筋肉痛 2480
関節痛 2480
悪心・
2480
嘔吐
悪寒 2480
初回免疫として本剤0.5mLの2回接種を完了した12〜17歳の者を対象に、2
回目接種から5ヵ月以上後に追加免疫として本剤0.25mLを1回接種したと
きの安全性、反応原性及び免疫原性を検討した。追加免疫として本剤
0.25mLを1回接種した372例のうち、追加免疫前のSARS‑CoV‑2検査結果
が陰性で免疫原性評価が規定どおり行われた327例を対象に接種後28日の
シュードウイルスに対する血清中和抗体濃度及び中和抗体応答率を評価
し、17.1.1海外第Ⅲ相試験の初回免疫後(2回目接種28日後)の成績と比
較した。結果は表10のとおりであった5)。
2回目
本剤群
プラセボ群
n(%)
n(%)
グレード 評価
グレード 評価
全体
全体
3以上a) 例数
3以上a) 例数
2310 133
431
1
1238
2478
(93.1)(5.4)
(34.8)(<0.1)
403
27
12
1238
0
2478
(16.2)(1.1)
(1.0)
578
10
101
1238
0
2477
(23.3)(0.4)
(8.2)
1106
56
477
17
1238
2478
(44.6)(2.3)
(38.5)(1.4)
1188
33
453
18
1238
2478
(47.9)(1.3)
(36.6)(1.5)
668
24
205
10
1238
2477
(26.9)(1.0)
(16.6)(0.8)
371
15
143
5
1238
2477
(15.0)(0.6)
(11.6)(0.4)
281
2
110
1238
0
2477
(11.3)(<0.1)
(8.9)
456
4
138
1
1238
2477
(18.4)(0.2)
(11.1)(<0.1)
本剤群
プラセボ群
n(%)
n(%)
グレード 評価
グレード
全体
全体
3以上a) 例数
3以上a)
2290 126
370
3
1220
(92.4)(5.1)
(30.3)(0.2)
509
56
12
1220
0
(20.5)(2.3)
(1.0)
519
7
61
1220
0
(21.0)(0.3)
(5.0)
1739 113
370
14
1220
(70.2)(4.6)
(30.3)(1.1)
1679 188
353
10
1220
(67.8)(7.6)
(28.9)(0.8)
1154 129
153
3
1220
(46.6)(5.2)
(12.5)(0.2)
716
57
113
2
1220
(28.9)(2.3)
(9.3)(0.2)
591
3
106
1220
0
(23.9)(0.1)
(8.7)
1066
11
97
1220
0
(43.0)(0.4)
(8.0)
表10 追加免疫時のシュードウイルスに対する血清中和抗体濃度及び中和
抗体応答率
12〜17歳
GMR
[両側95%CI]b,c)
N
N
(12〜17歳vs
血清中和
18〜25歳)
抗体濃度
7172.043
1400.411
5.121
257
294
[6535.156, 7870.999]
[1283.794, 1527.622][4.509, 5.817]
%
%
抗体応答率の差
n/N
n/N
e)
中和抗体
[両側95%CI]
[両側95%CI] [両側95%CI]
100
99.3
0.7
応答率d)
257/257
292/294
[98.6, 100.0]
[97.6, 99.9]
[‑0.8, 2.4]
17.1.4 海外第Ⅱa相試験(追加免疫)
SARS‑CoV‑2ワクチン未接種の18歳以上の者を対象に、初回免疫として
本剤0.25mL※又は0.5mLの2回接種を完了した後、2回目接種から6ヵ月以
上後に追加免疫として本剤0.25mLを1回接種したときの安全性、反応原性
及び免疫原性を検討した※※。初回免疫として本剤0.5mLの2回接種を完了
した後、追加免疫として本剤0.25mLを1回接種した171例のうち、追加免
疫前後の免疫原性評価が規定どおり行われた149例を対象に、追加免疫後
(接種28日後)のシュードウイルスに対する血清中和抗体価及び中和抗体
応答率を評価し、17.1.1海外第Ⅲ相試験の初回免疫後(2回目接種28日後)
の成績と比較した。結果は表8のとおりであった6)。
※初回免疫の承認用量は0.5mLである。
※※本試験の追加免疫後の免疫原性データについて、初回免疫時の2用量群
を併合したデータを用いて、起源株に対する中和抗体価のGMT及び抗体
応答率を主要評価項目とし、海外第Ⅲ相試験の初回免疫後の免疫原性デー
タに対する非劣性を検証する免疫ブリッジング解析を計画した。
安全性は、初回免疫として本剤0.5mLの2回接種を完了した後、追加免疫と
して本剤0.25mLを1回接種した1346例で評価した。接種後7日間は電子日誌
により副反応が収集され、接種後に発現頻度が20%を超えた副反応の発現
状況(全体及びグレード3以上)は表11のとおりであった。副反応の大部分
は、接種後1〜2日以内に発現し、持続期間中央値は2〜3日であった5)。
表11 主な副反応の発現状況
表8 追加免疫時のシュードウイルスに対する血清中和抗体価(50%阻害希
釈倍率)及び中和抗体応答率
GMR
[両側95%CI]b)
GLSMa,b)
GLSMa,b)
N
N
(追加免疫後vs
血清中和
[両側95%CI]
[両側95%CI]
初回免疫後)
抗体価
1802.426
1026.854
1.755
149
1053
[1548.020, 2098.643]
[967.880, 1089.420][1.496, 2.060]
%
%
抗体応答率の差
n/N
n/N
中和抗体
[両側95%CI]
[両側95%CI]
[両側95%CI]
87.9
98.4
‑10.5
応答率c)
131/149
1033/1050
[81.6, 92.7]
[97.4, 99.1] [‑16.7, ‑6.1]
安全性は、初回免疫として本剤0.5mLの2回接種を完了した後、追加免疫
として本剤0.25mLを1回接種した171例で評価した。接種後7日間は電子日
誌により副反応が収集され、接種後に発現頻度が20%を超えた副反応の発
現状況(全体及びグレード3以上)は表9のとおりであった。副反応の大部
分は、接種後1〜2日以内に発現し、持続期間中央値は1〜3日であった6)。
表9 主な副反応の発現状況
評価例数
注射部位疼痛
リンパ節症
頭痛
疲労
筋肉痛
関節痛
悪寒
167
167
167
167
167
167
167
評価例数
注射部位疼痛
リンパ節症
頭痛
疲労
筋肉痛
関節痛
悪寒
初回免疫後
N=評価例数、n=中和抗体応答がみられた被験者数
CI:信頼区間、GLSM:幾何最小二乗平均、GMR:幾何平均比
a)抗体価がLLOQ未満の場合、解析には0.5×LLOQの値が用いられた。
b)臨床試験(17.1.4海外第Ⅱa相試験、17.1.1海外第Ⅲ相試験)を固定効果、年齢
(65歳以上、65歳未満)を共変量としたANCOVA
c)抗体価がLLOQ未満からLLOQの4倍以上へ変化した被験者の割合又はベースライ
ンから4倍以上上昇した被験者の割合
全体
n(%)
140(83.8)
34(20.4)
92(55.1)
98(58.7)
82(49.1)
69(41.3)
59(35.3)
グレード3以上a)
n(%)
6(3.6)
1(0.6)
2(1.2)
7(4.2)
5(3.0)
5(3.0)
0
n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
GLSMa,b)
[両側95%CI]
N=評価例数、n=中和抗体応答がみられた被験者数
CI:信頼区間、GLSM:幾何最小二乗平均、GMR:幾何平均比
a)抗体価がLLOQ未満の場合、解析には0.5×LLOQの値が用いられ、抗体価が定量
上限(ULOQ)超の場合、ULOQの値が用いられた。
b)臨床試験(17.1.5海外第Ⅱ/Ⅲ相試験、17.1.1海外第Ⅲ相試験(18〜25歳))を固
定効果としたANCOVA
c)非劣性マージンは0.67(GMR(12〜17歳/18〜25歳)の両側95%CI下限>0.67)と
設定され、かつ試験の成功基準は点推定値が≥0.8とされた。
d)ベースライン時(初回免疫前)の抗体価がLLOQ未満であった場合はLLOQ未満か
らLLOQの4倍以上への変化、LLOQ以上であった場合はベースライン時抗体価の4
倍以上の上昇が得られた場合と定義した。
e)非劣性マージンは‑10%(抗体応答率の差(12〜17歳‑18〜25歳)の両側95%CI下限
>‑10%)と設定された。
n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
追加免疫後
18〜25歳
GLSMa,b)
[両側95%CI]
1294
1293
1293
1293
1293
1293
1293
全体
n(%)
1179(91.1)
363(28.1)
739(57.2)
759(58.7)
523(40.4)
311(24.1)
396(30.6)
グレード3以上a)
n(%)
38(2.9)
3(0.2)
28(2.2)
52(4.0)
44(3.4)
17(1.3)
7(0.5)
n=発現例数
a)重症度が「重度(日常生活を妨げる)」以上として報告された事象
18. 薬効薬理
18.1 作用機序
本剤は脂質ナノ粒子に封入されたヌクレオシド修飾メッセンジャーRNA
(mRNA)を含有する。脂質ナノ粒子によりmRNAは宿主細胞内に送達さ
れ、SARS‑CoV‑2のスパイクタンパク質を一過性に発現する。発現した
スパイクタンパク質は免疫細胞により外来抗原として認識され、これに対
する中和抗体産生及び細胞性免疫応答が誘導される。
20. 取扱い上の注意
20.1 外箱開封後は遮光して保存すること。
20.2 ‑50℃以下で保管しないこと。
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 現時点での知見が限られていることから、製造販売後、副反応情報等
の本剤の安全性に関するデータを、あらかじめ定めた計画に基づき早期に
収集するとともに、独立行政法人医薬品医療機器総合機構に提出し、本剤
の適正使用に必要な措置を講じること。その際、国が実施する健康調査等
により得られた情報についても適切に反映すること。
21.3 現在国内外で実施中又は計画中の臨床試験の成績が得られた際には、
速やかに当該成績を独立行政法人医薬品医療機器総合機構に提出するとと
もに、本剤の有効性及び安全性に係る最新の情報を、医療従事者及び被接
種者が容易に入手可能となるよう必要な措置を講じること。また、国が行
う本剤の有効性及び安全性に係る情報の発信について、適切に協力するこ
と。
21.4 本剤の接種に際し、本剤の有効性及び安全性については今後も情報が
集積されることを踏まえ、あらかじめ被接種者又は代諾者に最新の有効性
及び安全性に関する情報が文書をもって説明され、予診票等で文書による
同意を得てから接種されるよう、医師に対して適切に説明すること。
−4−