


よむ、つかう、まなぶ。
資料1-3:研究全体の責任主体の概念について (2 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_42147.html |
| 出典情報 | 厚生科学審議会 臨床研究部会(第35回 8/8)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
臨床研究法施行5年後の見直しに係る検討のとりまとめ(概要)
革新的な医薬品等の研究開発の推進
1.臨床研究実施体制の国際整合性
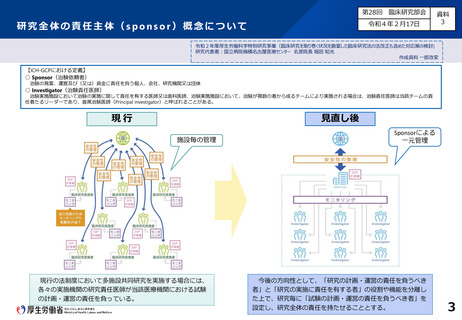
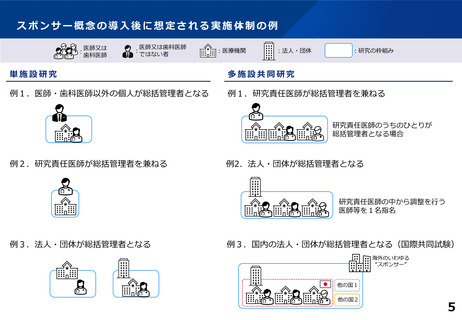
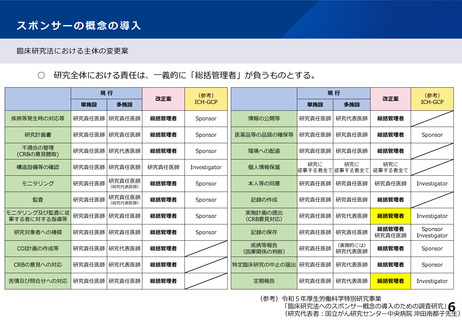
(1)研究全体の責任主体の概念について
■ 国際整合性の観点も踏まえ、多施設共同研究においても統一的な運営体制が確保さ
れるよう、臨床研究の実施体制について「研究の計画・運営の責任を負うべき者」と「研究
の実施に責任を有する者」の役割や機能を整理した上で、研究毎に研究の実施を統一
的に管理する 「試験の計画・運営の責任を負うべき者」を設定すべき。
■ 再審査・再評価に係る製造販売後臨床試験以外の製造販売後の臨床試験について
は、薬機法下の適切な基準に準拠して実施することができるようにすべき。
(2)特定臨床研究で得られた情報の薬事申請における利活用について
■ 厚生労働科学特別研究事業等において、特定臨床研究で得られたデータを薬事申請
に活用する場合の一般的な要件、留意点等の取りまとめ・公表に向けた検討を進めるべ
き。
(3)いわゆる観察研究に関する臨床研究法の適用範囲について
※令和4年6月3日厚生科学審議会臨床研究部会とりまとめ
3.手続の合理化
(1)届出・変更手続きの合理化、届出のオンライン化について
■ 現行法において、変更の届出が必要な項目のうち、研究の本質に関わらないような事
項は軽微変更とすべき。
■ 届出事項としなくても、jRCTに掲載し、公開できれば良い項目を整理し、実施計画と
jRCTへの掲載項目を分離すべき。
■ 届出のオンライン化、jRCTの改修に着手すべき。
(2)利益相反申告手続の適正化について
■ COI管理について、医療機関における事実確認の手続を代替するための客観的かつ容
易な確認や、臨床研究法における特定臨床研究のみならず国内の医学系研究に関する
COIの一元管理が可能となるようなデータベースを構築することが望ましい。
■ 国がこのようなデータベースの構築に向けた取組に着手することが期待される。
研究の信頼性確保
■ 法の対象となる臨床研究の範囲は、研究計画に従って研究対象者に対し医薬品等を
使用する研究及び適切な医療として医薬品等を使用するものであって、研究対象者への 1.透明性の確保
通常の医療と大きく異なる傷害・負担が大きい検査等を研究目的で診療に追加して行う (1)利益相反申告手続の適正化について(再掲)
研究とすべき。
(2)研究資金等の提供に関する情報公表の範囲について
■ 「傷害・負担が大きい検査等」の基準や事例を明示すべきであり、引き続き、事例の収
■ 特定臨床研究に関与する企業について、費目の付け替えが行われている可能性の
集や基準に係る考え方の検討を進めるべき。
有無を確認できる状態とするよう、企業における情報提供関連費及び接遇費の年
(4)疾病等報告の取扱いについて
間総額の公表を法令で義務付けるべきである。
■ 研究毎に設定される「試験の計画・運営の責任を負うべき者」において、有害事象に係 (3)重大な不適合の取扱いについて
る情報を一元的に集約し、因果関係について一律に判断できるようにすべき。
■ 特定臨床研究において、重大な不適合が発生した場合には、研究機関の長に
■ 未承認・適応外の医薬品等の臨床研究における既知の重篤な疾病等をCRBに報告
公表を求めることとする。
する期限については、原則30日以内とすることとし、研究組織から独立した効果安全性
2.研究の質の確保
評価委員会が設置される場合には、その運用を示した上で定期報告とすべき。
■ 既承認の医薬品等の臨床研究については、通常の診療においても起こりうる事象であり、 (1)臨床研究審査委員会の認定要件について
既知の疾病等をCRBに報告する期限は定期とすべき。
■ 更新要件については、これまでの開催回数の要件を見直すとともに、新規の審議件
数を要件に加えるべき。
2.研究の法への該当性の明確化
■ 当面、新規の審議件数は3年間で6件以上(ただし、毎年1件以上)、かつ、
(1)適応外薬に関する特定臨床研究の対象範囲について
開催回数については毎年7回以上とする。ただし、疾病等報告等、迅速に取り扱う
■ 適応外医薬品等を使用する研究であっても、既承認の用法等とリスクが同程度のもの
議題がある場合には、要件に関わらず、迅速な開催を求めることとする。
については、特定臨床研究の範囲から除外することとし、リスクの判断にあたっては、臨床
■ 要件を満たさない場合は、廃止に向けた円滑な準備を進めていただくこととする。
研究部会の下に専門委員会を設置して検討すべき。
■ 今後、定期的にCRBの活動状況を確認しそれらを分析した上で、必要な見直しを
(2)医療機器に関する臨床研究法の適用範囲について
行っていく。
■ 医療機器を用いた研究に関し法への該当性等を相談できるよう、相談窓口の設置を
※ CRBの更新要件について省令改正を行い、一定の経過措置を設けた上で、新規の審議
進めるべき。
件数は3年間で6件以上(ただし、毎年1件以上)、かつ、開催回数については毎年
■ 定期的に特定臨床研究の該当判断に迷った事例等の収集を行い、随時事例集を更
7以上に見直しを行った。
新していくべき。
※ 更新要件を満たさないCRBについては、円滑な廃止に向けて準備を進めていただくものとし、
■ 関係学会等の協力を得て、臨床研究法に関するQ&Aや事例集をCRB、倫理審査委
これまでに発出したCRBの更新に係る事務連絡を廃止した上で、改めてCRBの更新に
関する考え方を示した。
員会、工学部の研究者等を含めた関係者に広く周知していくべき。
2
革新的な医薬品等の研究開発の推進
1.臨床研究実施体制の国際整合性
(1)研究全体の責任主体の概念について
■ 国際整合性の観点も踏まえ、多施設共同研究においても統一的な運営体制が確保さ
れるよう、臨床研究の実施体制について「研究の計画・運営の責任を負うべき者」と「研究
の実施に責任を有する者」の役割や機能を整理した上で、研究毎に研究の実施を統一
的に管理する 「試験の計画・運営の責任を負うべき者」を設定すべき。
■ 再審査・再評価に係る製造販売後臨床試験以外の製造販売後の臨床試験について
は、薬機法下の適切な基準に準拠して実施することができるようにすべき。
(2)特定臨床研究で得られた情報の薬事申請における利活用について
■ 厚生労働科学特別研究事業等において、特定臨床研究で得られたデータを薬事申請
に活用する場合の一般的な要件、留意点等の取りまとめ・公表に向けた検討を進めるべ
き。
(3)いわゆる観察研究に関する臨床研究法の適用範囲について
※令和4年6月3日厚生科学審議会臨床研究部会とりまとめ
3.手続の合理化
(1)届出・変更手続きの合理化、届出のオンライン化について
■ 現行法において、変更の届出が必要な項目のうち、研究の本質に関わらないような事
項は軽微変更とすべき。
■ 届出事項としなくても、jRCTに掲載し、公開できれば良い項目を整理し、実施計画と
jRCTへの掲載項目を分離すべき。
■ 届出のオンライン化、jRCTの改修に着手すべき。
(2)利益相反申告手続の適正化について
■ COI管理について、医療機関における事実確認の手続を代替するための客観的かつ容
易な確認や、臨床研究法における特定臨床研究のみならず国内の医学系研究に関する
COIの一元管理が可能となるようなデータベースを構築することが望ましい。
■ 国がこのようなデータベースの構築に向けた取組に着手することが期待される。
研究の信頼性確保
■ 法の対象となる臨床研究の範囲は、研究計画に従って研究対象者に対し医薬品等を
使用する研究及び適切な医療として医薬品等を使用するものであって、研究対象者への 1.透明性の確保
通常の医療と大きく異なる傷害・負担が大きい検査等を研究目的で診療に追加して行う (1)利益相反申告手続の適正化について(再掲)
研究とすべき。
(2)研究資金等の提供に関する情報公表の範囲について
■ 「傷害・負担が大きい検査等」の基準や事例を明示すべきであり、引き続き、事例の収
■ 特定臨床研究に関与する企業について、費目の付け替えが行われている可能性の
集や基準に係る考え方の検討を進めるべき。
有無を確認できる状態とするよう、企業における情報提供関連費及び接遇費の年
(4)疾病等報告の取扱いについて
間総額の公表を法令で義務付けるべきである。
■ 研究毎に設定される「試験の計画・運営の責任を負うべき者」において、有害事象に係 (3)重大な不適合の取扱いについて
る情報を一元的に集約し、因果関係について一律に判断できるようにすべき。
■ 特定臨床研究において、重大な不適合が発生した場合には、研究機関の長に
■ 未承認・適応外の医薬品等の臨床研究における既知の重篤な疾病等をCRBに報告
公表を求めることとする。
する期限については、原則30日以内とすることとし、研究組織から独立した効果安全性
2.研究の質の確保
評価委員会が設置される場合には、その運用を示した上で定期報告とすべき。
■ 既承認の医薬品等の臨床研究については、通常の診療においても起こりうる事象であり、 (1)臨床研究審査委員会の認定要件について
既知の疾病等をCRBに報告する期限は定期とすべき。
■ 更新要件については、これまでの開催回数の要件を見直すとともに、新規の審議件
数を要件に加えるべき。
2.研究の法への該当性の明確化
■ 当面、新規の審議件数は3年間で6件以上(ただし、毎年1件以上)、かつ、
(1)適応外薬に関する特定臨床研究の対象範囲について
開催回数については毎年7回以上とする。ただし、疾病等報告等、迅速に取り扱う
■ 適応外医薬品等を使用する研究であっても、既承認の用法等とリスクが同程度のもの
議題がある場合には、要件に関わらず、迅速な開催を求めることとする。
については、特定臨床研究の範囲から除外することとし、リスクの判断にあたっては、臨床
■ 要件を満たさない場合は、廃止に向けた円滑な準備を進めていただくこととする。
研究部会の下に専門委員会を設置して検討すべき。
■ 今後、定期的にCRBの活動状況を確認しそれらを分析した上で、必要な見直しを
(2)医療機器に関する臨床研究法の適用範囲について
行っていく。
■ 医療機器を用いた研究に関し法への該当性等を相談できるよう、相談窓口の設置を
※ CRBの更新要件について省令改正を行い、一定の経過措置を設けた上で、新規の審議
進めるべき。
件数は3年間で6件以上(ただし、毎年1件以上)、かつ、開催回数については毎年
■ 定期的に特定臨床研究の該当判断に迷った事例等の収集を行い、随時事例集を更
7以上に見直しを行った。
新していくべき。
※ 更新要件を満たさないCRBについては、円滑な廃止に向けて準備を進めていただくものとし、
■ 関係学会等の協力を得て、臨床研究法に関するQ&Aや事例集をCRB、倫理審査委
これまでに発出したCRBの更新に係る事務連絡を廃止した上で、改めてCRBの更新に
関する考え方を示した。
員会、工学部の研究者等を含めた関係者に広く周知していくべき。
2