



よむ、つかう、まなぶ。
試験の概要「ヌバキソビッド筋注」 (3 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_26943.html |
| 出典情報 | 薬事・食品衛生審議会 医薬品第二部会 資料(7/20)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
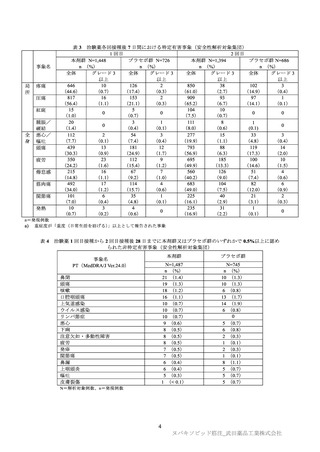
表2
小児拡大パート(12~17 歳)及びメインスタディ(18~25 歳)における 2 回目接種後 14 日の
従来株に対する中和抗体価の GMT の比及び SCR の差(PP 免疫原性解析対象集団)
12~17歳
(小児拡大パート)
18~25歳
(メインスタディ)
N
390
416
GMT a)[両側95%CI]
3859.6[3422.8, 4352.1]
2633.6[2388.6, 2903.6]
GMR b)[両側95%CI]
0.7[0.6, 0.8]
n/N
385/390
415/416
SCR c) %[両側95%CI]
98.7[97.0, 99.6]
99.8[98.7, 100.0]
SCRの差[両側95%CI]
d)
1.1[-0.2, 2.8]
CI:信頼区間、GMR:幾何平均比、GMT:幾何平均、N:評価例数、n:抗体陽転がみられた被験者数
a) 抗体価が定量下限値(LLOQ)未満の場合、解析には 0.5×LLOQ の値を用いた。
b) 接種群を要因、ベースラインの抗体価を共変量とした ANCOVA
c) 抗体価が 1 回目接種日から 4 倍以上増加した被験者の割合
d) Miettinen and Nurminen 法
安全性について、観察期間は以下のとおりとされた。
特定有害事象 4)(局所(疼痛、圧痛、紅斑及び腫脹/硬結)及び全身(悪心/嘔吐、頭
痛、疲労、倦怠感、筋肉痛、関節痛及び発熱)):初回評価期間(クロスオーバー前)
の治験薬の各回接種後 7 日間5)(被験者日誌により収集された)
すべての非特定有害事象:初回評価期間とクロスオーバー期間それぞれの治験薬 1 回目
接種から 2 回目接種後 28 日後まで
診療を要した有害事象:初回評価期間とクロスオーバー期間それぞれの治験薬 1 回目接
種から 2 回目接種後 28 日後まで(うち治験薬接種との因果関係ありとされた事象は初
回評価期間の治験薬 2 回目接種後 720 日間)
重篤な有害事象及び注目すべき有害事象:同意取得から初回評価期間の治験薬 2 回目接
種後 720 日間
安全性解析対象集団において、初回評価期間の治験薬各回接種後 7 日間に発現した特定有
害事象を表 3 に示す。本剤群では 1 回目接種後より 2 回目接種後に特定有害事象の発現割合
が増加した。特定有害事象の重症度は多くがグレード 1 又は 2 であった。本剤群でみられた
特定有害事象の大部分は接種後 1~2 日以内に発現し、持続期間の中央値は 1~2 日であった。
安全性解析対象集団において、1 回目接種後 49 日までの非特定有害事象の発現割合は、本
剤群 16.2%(241/1,487 例)及びプラセボ群 15.7%(117/745 例)であり、うち、治験薬接種と
の因果関係が否定されなかった非特定有害事象の発現割合は本剤群 3.4%(51/1,487 例)及び
プラセボ群 1.1%(8/745 例)であった。いずれかの接種群で 0.5%以上に発現した非特定有害
事象は表 4 のとおりであり、そのうち治験薬接種との因果関係の否定されなかった非特定有
害事象はリンパ節症(本剤群 0.5%(7/1,487 例)、プラセボ群 0%(0/745 例))であった。
特定有害事象の重症度は予防ワクチンの臨床試験における毒性評価尺度に関する FDA ガイダンス(Guidance for Industry
Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials, 2007 年 9
月)に基づき評価された。
5)
接種から 7 日後以降までグレード 1 以上が継続した事象については、7 日後以降は接種後 7 日目を起点日とした非特
定有害事象として事象が消失するまで収集された。
4)
3
ヌバキソビッド筋注_武田薬品工業株式会社
小児拡大パート(12~17 歳)及びメインスタディ(18~25 歳)における 2 回目接種後 14 日の
従来株に対する中和抗体価の GMT の比及び SCR の差(PP 免疫原性解析対象集団)
12~17歳
(小児拡大パート)
18~25歳
(メインスタディ)
N
390
416
GMT a)[両側95%CI]
3859.6[3422.8, 4352.1]
2633.6[2388.6, 2903.6]
GMR b)[両側95%CI]
0.7[0.6, 0.8]
n/N
385/390
415/416
SCR c) %[両側95%CI]
98.7[97.0, 99.6]
99.8[98.7, 100.0]
SCRの差[両側95%CI]
d)
1.1[-0.2, 2.8]
CI:信頼区間、GMR:幾何平均比、GMT:幾何平均、N:評価例数、n:抗体陽転がみられた被験者数
a) 抗体価が定量下限値(LLOQ)未満の場合、解析には 0.5×LLOQ の値を用いた。
b) 接種群を要因、ベースラインの抗体価を共変量とした ANCOVA
c) 抗体価が 1 回目接種日から 4 倍以上増加した被験者の割合
d) Miettinen and Nurminen 法
安全性について、観察期間は以下のとおりとされた。
特定有害事象 4)(局所(疼痛、圧痛、紅斑及び腫脹/硬結)及び全身(悪心/嘔吐、頭
痛、疲労、倦怠感、筋肉痛、関節痛及び発熱)):初回評価期間(クロスオーバー前)
の治験薬の各回接種後 7 日間5)(被験者日誌により収集された)
すべての非特定有害事象:初回評価期間とクロスオーバー期間それぞれの治験薬 1 回目
接種から 2 回目接種後 28 日後まで
診療を要した有害事象:初回評価期間とクロスオーバー期間それぞれの治験薬 1 回目接
種から 2 回目接種後 28 日後まで(うち治験薬接種との因果関係ありとされた事象は初
回評価期間の治験薬 2 回目接種後 720 日間)
重篤な有害事象及び注目すべき有害事象:同意取得から初回評価期間の治験薬 2 回目接
種後 720 日間
安全性解析対象集団において、初回評価期間の治験薬各回接種後 7 日間に発現した特定有
害事象を表 3 に示す。本剤群では 1 回目接種後より 2 回目接種後に特定有害事象の発現割合
が増加した。特定有害事象の重症度は多くがグレード 1 又は 2 であった。本剤群でみられた
特定有害事象の大部分は接種後 1~2 日以内に発現し、持続期間の中央値は 1~2 日であった。
安全性解析対象集団において、1 回目接種後 49 日までの非特定有害事象の発現割合は、本
剤群 16.2%(241/1,487 例)及びプラセボ群 15.7%(117/745 例)であり、うち、治験薬接種と
の因果関係が否定されなかった非特定有害事象の発現割合は本剤群 3.4%(51/1,487 例)及び
プラセボ群 1.1%(8/745 例)であった。いずれかの接種群で 0.5%以上に発現した非特定有害
事象は表 4 のとおりであり、そのうち治験薬接種との因果関係の否定されなかった非特定有
害事象はリンパ節症(本剤群 0.5%(7/1,487 例)、プラセボ群 0%(0/745 例))であった。
特定有害事象の重症度は予防ワクチンの臨床試験における毒性評価尺度に関する FDA ガイダンス(Guidance for Industry
Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials, 2007 年 9
月)に基づき評価された。
5)
接種から 7 日後以降までグレード 1 以上が継続した事象については、7 日後以降は接種後 7 日目を起点日とした非特
定有害事象として事象が消失するまで収集された。
4)
3
ヌバキソビッド筋注_武田薬品工業株式会社