













よむ、つかう、まなぶ。
資料1:臨床研究法の見直しに係る各論点について (5 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_24643.html |
| 出典情報 | 厚生科学審議会 臨床研究部会(第29回 3/24)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
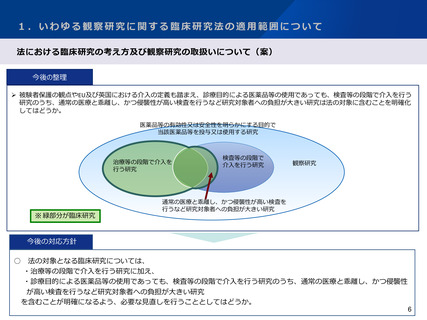
1.いわゆる観察研究に関する臨床研究法の適用範囲について
(参考)海外等における観察研究の取扱い
参照国/
規制
令和3年度厚生労働科学特別研究事業(臨床研究法見直し審議における新たな課題・論点への対応策の確立のための研究)
研究代表者:国立病院機構名古屋医療センター 名誉院長 堀田 知光
作成資料 一部改変
サマリ
• 連邦助成を受ける人を対象とするすべての研究は、介入の有無によらず被験者保護の基本規則であるコモン・ルール(45 CFR
46)の適用を受ける。
米国
• 医薬品を用いた臨床試験を規制する21 CFR 312におけるIND申請では、日常診療どおりに医薬品が用いられる非介入研究は対
象外である。
• 医療機器を用いた臨床試験を行う際にFDAに対して必要なIDE申請は市販前の医療機器を対象としており、市販の医療機器が用
いられる非介入研究は対象外である。
EU
英国
• 医薬品に関するEU規制(2014/536; EU-CTR)は、非介入研究には適用されない。ただし、EU-CTRでの介入の定義には、医薬品
だけではなく日常診療を超える診断やモニタリングの上乗せも含まれ、その目的が有効性・安全性等を明らかにすることであ
ればEU-CTRの対象となり得る。ただし、こうした診断やモニタリングの介入の多くは低介入臨床試験(low-interventional
clinical trial)に分類され、その場合には、モニタリングや医薬品管理、必須文書の内容、補償等の義務が大幅に緩和される。
•
医療機器に関するEU規制(2017/745; EU-MDR)は、医療機器及びその付属品の上市や使用開始に関する規則であり、医療機
器の安全性または性能を評価するために行われるヒトを対象とした研究に広く適用される。植込み機器およびクラスIII機器で
は、原則、臨床研究(clinical investigation)を実施しなければならず、全般的にクラス分類や植込み機器かどうかによる規定
の場合分けがなされており、介入の有無によって規制の該当性は区別されていない。
• 英国における医薬品に関する規制であるThe Medicines for Human Use (Clinical Trials) Regulations 2004では非介入研究(noninterventional clinical trial)は上記の規制の対象外とされている。ただし、介入の定義はEU-CTRと同様の考え方であり、日常
診療を超える診断やモニタリングの上乗せが行われ、その目的が有効性・安全性等を明らかにすることであれば規制の対象と
なり得る。
• 現在の英国の医薬品規制はEU Directive (2001/20)をベースにしたものであり、新たなEU regulation(536/2014)の施行にあわ
せて、規制要件の調和が図られる見通しである。
• 英国における医療機器の規制は、今後EU-MDR等の国際的な規制要件への調和が図られる見通しである。
ICHE6/E8
• ICH-E6(ICH-GCP)、ICH-E8(臨床試験の一般原則)の対象は医薬品を用いる介入研究(interventional studies for medical
products)であり、非介入研究は対象外である。
5
(参考)海外等における観察研究の取扱い
参照国/
規制
令和3年度厚生労働科学特別研究事業(臨床研究法見直し審議における新たな課題・論点への対応策の確立のための研究)
研究代表者:国立病院機構名古屋医療センター 名誉院長 堀田 知光
作成資料 一部改変
サマリ
• 連邦助成を受ける人を対象とするすべての研究は、介入の有無によらず被験者保護の基本規則であるコモン・ルール(45 CFR
46)の適用を受ける。
米国
• 医薬品を用いた臨床試験を規制する21 CFR 312におけるIND申請では、日常診療どおりに医薬品が用いられる非介入研究は対
象外である。
• 医療機器を用いた臨床試験を行う際にFDAに対して必要なIDE申請は市販前の医療機器を対象としており、市販の医療機器が用
いられる非介入研究は対象外である。
EU
英国
• 医薬品に関するEU規制(2014/536; EU-CTR)は、非介入研究には適用されない。ただし、EU-CTRでの介入の定義には、医薬品
だけではなく日常診療を超える診断やモニタリングの上乗せも含まれ、その目的が有効性・安全性等を明らかにすることであ
ればEU-CTRの対象となり得る。ただし、こうした診断やモニタリングの介入の多くは低介入臨床試験(low-interventional
clinical trial)に分類され、その場合には、モニタリングや医薬品管理、必須文書の内容、補償等の義務が大幅に緩和される。
•
医療機器に関するEU規制(2017/745; EU-MDR)は、医療機器及びその付属品の上市や使用開始に関する規則であり、医療機
器の安全性または性能を評価するために行われるヒトを対象とした研究に広く適用される。植込み機器およびクラスIII機器で
は、原則、臨床研究(clinical investigation)を実施しなければならず、全般的にクラス分類や植込み機器かどうかによる規定
の場合分けがなされており、介入の有無によって規制の該当性は区別されていない。
• 英国における医薬品に関する規制であるThe Medicines for Human Use (Clinical Trials) Regulations 2004では非介入研究(noninterventional clinical trial)は上記の規制の対象外とされている。ただし、介入の定義はEU-CTRと同様の考え方であり、日常
診療を超える診断やモニタリングの上乗せが行われ、その目的が有効性・安全性等を明らかにすることであれば規制の対象と
なり得る。
• 現在の英国の医薬品規制はEU Directive (2001/20)をベースにしたものであり、新たなEU regulation(536/2014)の施行にあわ
せて、規制要件の調和が図られる見通しである。
• 英国における医療機器の規制は、今後EU-MDR等の国際的な規制要件への調和が図られる見通しである。
ICHE6/E8
• ICH-E6(ICH-GCP)、ICH-E8(臨床試験の一般原則)の対象は医薬品を用いる介入研究(interventional studies for medical
products)であり、非介入研究は対象外である。
5