







よむ、つかう、まなぶ。
○1 患者申出療養の中間報告について別紙2 (11 ページ)
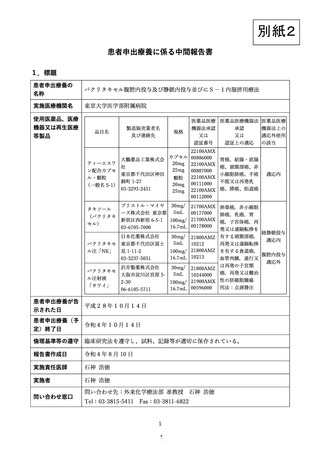
出典
| 公開元URL | https://www.mhlw.go.jp/stf/shingi2/0000203222_00020.html |
| 出典情報 | 患者申出療養評価会議(第33回 9/22)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
考察:
腹膜播種陽性または腹腔洗浄細胞診陽性の胃癌症例を対象として、S-1+パクリタキセル経静脈・
腹腔内併用療法の安全性と有効性を評価するための臨床研究を実施した。試験開始より5年時点の
中間解析結果を基に、本治療の安全性および有効性について、先進医療制度下に実施した先行研究
(P1症例対象 第Ⅲ相試験、P0CY1症例対象 第Ⅱ相試験)の結果を参考に考察する。
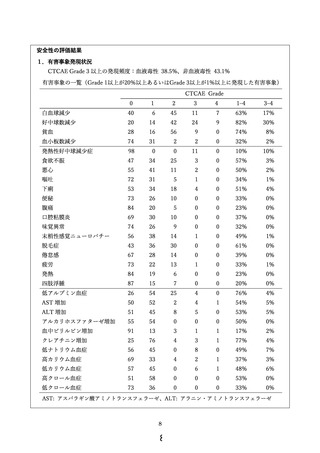
主要評価項目である有害事象発現状況に関しては、未知の重篤な有害事象および治療関連死亡を
認めず、主な有害事象および腹腔ポート関連合併症の発現頻度は下表の通りであった。
対象
n
白血球
減少
好中球
数減少
貧血
発熱性
好中球
減少症
下痢
食欲
不振
ポート
関連
合併症
本研究
P1/CY1
109
17%
30%
8%
10%
4%
3%
10%
第Ⅲ相試験
P1
116
25%
50%
13%
8%
9%
10%
6%
第Ⅱ相試験
P0CY1
38
11%
34%
13%
0%
5%
5%
5%
試験
本研究において、本療法で頻度が高く、治療の忍容性に影響する好中球数減少を抑えられたこと
は、治療の継続性と有効性につながったものと推定される。本研究では、患者申出療養制度の趣旨
に基づき、先行研究では対象外であった高齢者(75歳以上85歳未満)
、PS 2症例、2か月以上の前治
療歴を有する症例等も登録可能とし、安全性確保のため担当医判断による投与量の調整を許容した。
その結果として有害事象の発現を抑えることができたものと考えられる。
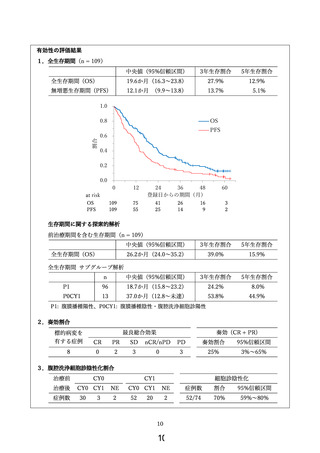
副次評価項目である有効性に関しては、下表の通りの結果が得られた。
試験
本研究
対象
n
全生存期間
腹腔洗浄細胞診陰性化割合
P1
96
18.7か月(15.8~23.2)
P0CY1
13
37.0か月(12.8~未達)
114
17.7か月(14.7~21.5)
76% (67%~85%)
38
27.5か月(21.9~36.9)
95% (82%~99%)
第Ⅲ相試験
P1
第Ⅱ相試験
P0CY1
70% (59%~80%)
本研究は先行研究と適格基準が異なり、実際に登録された症例の背景が異なるため、結果の比較
可能性はない。
※
先行する第Ⅲ相試験において、本療法の標準化学療法に対する優越性は示されなかった(生存期
間中央値 17.7か月対15.2か月、p=0.080、ハザード比 0.72 [95% CI 0.49–1.04])ものの、実施計画
書に適合した対象集団における探索的解析、腹水量の不均衡を調整した解析および追跡調査では、
本療法の臨床的な有効性を示唆する結果が得られている。
結論:中間解析により先行研究と同様の結果が得られたことより、S-1+パクリタキセル経静脈・
腹腔内併用療法は安全に実施可能であり、有効であることが示唆された。
報告書作成日:令和4年8月10日
11
11
腹膜播種陽性または腹腔洗浄細胞診陽性の胃癌症例を対象として、S-1+パクリタキセル経静脈・
腹腔内併用療法の安全性と有効性を評価するための臨床研究を実施した。試験開始より5年時点の
中間解析結果を基に、本治療の安全性および有効性について、先進医療制度下に実施した先行研究
(P1症例対象 第Ⅲ相試験、P0CY1症例対象 第Ⅱ相試験)の結果を参考に考察する。
主要評価項目である有害事象発現状況に関しては、未知の重篤な有害事象および治療関連死亡を
認めず、主な有害事象および腹腔ポート関連合併症の発現頻度は下表の通りであった。
対象
n
白血球
減少
好中球
数減少
貧血
発熱性
好中球
減少症
下痢
食欲
不振
ポート
関連
合併症
本研究
P1/CY1
109
17%
30%
8%
10%
4%
3%
10%
第Ⅲ相試験
P1
116
25%
50%
13%
8%
9%
10%
6%
第Ⅱ相試験
P0CY1
38
11%
34%
13%
0%
5%
5%
5%
試験
本研究において、本療法で頻度が高く、治療の忍容性に影響する好中球数減少を抑えられたこと
は、治療の継続性と有効性につながったものと推定される。本研究では、患者申出療養制度の趣旨
に基づき、先行研究では対象外であった高齢者(75歳以上85歳未満)
、PS 2症例、2か月以上の前治
療歴を有する症例等も登録可能とし、安全性確保のため担当医判断による投与量の調整を許容した。
その結果として有害事象の発現を抑えることができたものと考えられる。
副次評価項目である有効性に関しては、下表の通りの結果が得られた。
試験
本研究
対象
n
全生存期間
腹腔洗浄細胞診陰性化割合
P1
96
18.7か月(15.8~23.2)
P0CY1
13
37.0か月(12.8~未達)
114
17.7か月(14.7~21.5)
76% (67%~85%)
38
27.5か月(21.9~36.9)
95% (82%~99%)
第Ⅲ相試験
P1
第Ⅱ相試験
P0CY1
70% (59%~80%)
本研究は先行研究と適格基準が異なり、実際に登録された症例の背景が異なるため、結果の比較
可能性はない。
※
先行する第Ⅲ相試験において、本療法の標準化学療法に対する優越性は示されなかった(生存期
間中央値 17.7か月対15.2か月、p=0.080、ハザード比 0.72 [95% CI 0.49–1.04])ものの、実施計画
書に適合した対象集団における探索的解析、腹水量の不均衡を調整した解析および追跡調査では、
本療法の臨床的な有効性を示唆する結果が得られている。
結論:中間解析により先行研究と同様の結果が得られたことより、S-1+パクリタキセル経静脈・
腹腔内併用療法は安全に実施可能であり、有効であることが示唆された。
報告書作成日:令和4年8月10日
11
11