










よむ、つかう、まなぶ。
資料4-1 感染症定期報告感染症別文献一覧表[476KB] (7 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_44308.html |
| 出典情報 | 薬事審議会 医薬品等安全対策部会(令和6年度第2回 10/24)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
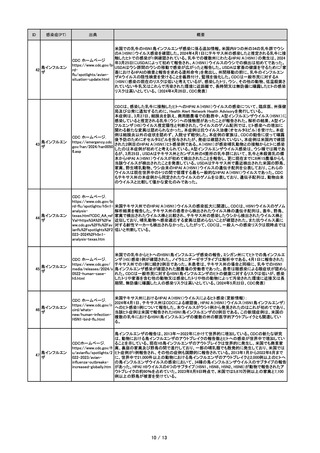
ID
感染症(PT)
出典
概要
ロタウイルス感
染
従来とは異なるG3P[6]遺伝子型を有するヒトロタウイルス株は、世界各地の下痢症患者から散発的
に検出されている。しかし、全ゲノムの塩基配列が決定され、特徴が明らかにされているアジア諸国
(中国、インドネシア、ベトナム)由来のヒトG3P[6]株は3株のみであることから、アジアにおける
G3P[6]株の正確な起源と進化は未だ解明されていない。本報告では、日本(千葉)において、2020
年12月に急性胃腸炎で入院した3カ月齢の男児の便検体から検出されたG3P[6]株(RVA/Humanwt/JPN/SO1199/2020/G3P[6])の全ゲノムの配列を決定し、特徴付けを行った。完全ゲノム解析
で、SO1199株は独特のWa様遺伝子群配列:G3-P[6]-I5-R1-C1-M1-A8-N1-T1-E1-H1を有するこ
とが明らかになった。VP6遺伝子型I5及びNSP1遺伝子型A8は、ブタロタウイルス株において一般的
に認められる。さらに、系統解析により、SO1199株の11遺伝子はすべて、ブタ又はブタ様ヒトロタウ
Infection, Genetics and
イルスの遺伝子と密接に関連しており、したがってブタ由来であることが示唆された。このように、
Evolution.
SO1199株はブタ様遺伝子のバックボーンを保有していることが示されたことから、ブタロタウイルス
115(2023)105507
株の種間伝播の結果である可能性が高い。SO1199株の全11遺伝子が、これまでに同定されたアジ
アを含む世界中のブタ様ヒトG3P[6]株とは異なるクラスター内に系統発生学的に位置しており、ブタ
からヒトへの独立した人畜共通感染症の発生が示唆された。SO1199株がブタ由来であることは、ヒト
とブタなどの家畜との間に近接することによる人獣共通感染症の発生を示唆している。実際、
SO1199株が検出された千葉県はブタが多く、ブタとヒトが密接に接触している一大畜産地である。し
たがって、SO1199株が検出された男児は直接的あるいは間接的にブタと接触していた可能性があ
る。しかし、SO1199株の遺伝子について、ブタ株よりブタ様ヒト株の方が配列同一性が高く、SO1199
株の進化経路はブタからの直接的な種間伝播よりも複雑かもしれず、ヒトと動物の両方でロタウイル
ス株を調査する必要がある。
ロタウイルス感
染
本研究では、ケニアの下痢患児(生後48カ月の女児)の便検体から検出されたG4P[6]株(RVA
(Group A rotavirus)/Human-wt/KEN/KCH148/2019/G4P[6])の全ゲノムの特性を評価した。完全
ゲノム解析により、KCH148株はWa様遺伝子群配列:G4-P[6]-I1-R1-C1-M1-A1-N1-T7-E1-H1を
有することが判明した。NSP3遺伝子型T7は、ブタロタウイルス株において一般的に認められ、系統
Infection, Genetics and 解析の結果、KCH148株の11遺伝子のうち10遺伝子がブタ由来であり、残りのNSP2遺伝子はヒト由
Evolution.
来であることが示された。KCH148株はブタロタウイルスの骨格を有しており、ブタ由来である可能性
96(2021)105133
が高いことが判明した。また、KCH148株がブタ由来であることは、ヒトと家畜が近接していることに起
因する人獣共通感染も示唆しており、特にヒトとブタなどの家畜との密接な接触があるアフリカの発
展途上国で顕著である。実際、KCH148株が発見されたKiambu郡は、ヒトと家畜が密接に接触してい
る農村部を有する都市周辺の郡であり、KCH148株が検出された患児がブタと接触していた可能性
がある。
ロタウイルス感
染
Virus Genes.
57(2021)338-357
レオウイルス科に属するA群ロタウイルス(RVA)は、ヒト及び世界中の多くの動物種の双方において
重度の胃腸炎の重要な病原体である。ほとんどのRVAは宿主に限定されているようであるが、再集
合を伴う又は伴わないRVAの種間伝播が観察されている。ヒトRVAsに関しては、6G(G1~G4、G9、
G12)と3P( P[4]、P[6]、P[8])が主要な遺伝子型と考えられているが、ブタRVAsはほとんどが5G(G3
~G5、G9、G11)と2P(P[6]、P[7])の遺伝子型に割り当てられている。ヒトではG4は通常P[8]遺伝子
型と関連しているが、ブタではP[6]と結合したG4が主に検出される。一方、世界各地の下痢症患者
では従来とは異なるG4P[6]遺伝子型が散発的に検出されている。これらのG4P[6]株のいくつかの全
ゲノムの塩基配列が決定され、特徴が明らかにされたことから、これらのヒト株の大部分がブタ由来
であることを示す証拠が得られた。しかし、ヒトG4P[6]株の正確な進化パターンは、独特で菌株特異
的な遺伝子型群を保有しているため、未だ解明されていない。本研究では、2014年と2015年に、タイ
の重症下痢症入院患者において、2つのG4P[6]株、DU2014-259とPK2015-1-0001を同定し、計514
のRVA陽性糞便検体をG/P遺伝子型タイピングにより検査した。結果、2株ともG4-P[6]-I1-R1-C1M1-A8-N1-T1-E1-H1という独特のWa様遺伝子型群を示した。NSP1遺伝子型A8はブタロタウイル
ス株で一般的に見られる遺伝子型である。さらに、系統解析の結果、DU2014-259株及びPK2015-10001株の11遺伝子はいずれもブタ由来と考えられた。一方、11遺伝子セグメントのうち9遺伝子セグ
メント(VP4、VP6、VP1~VP3、NSP2~NSP5)において一貫して異なるクラスターを形成しており、ブ
タからヒトへの独立した種間伝播が起こっていることが強く示唆された。この結果は、人獣共通感染
症であるG4P[6]株の起源や、ブタとヒトのロタウイルス株の動的相互作用に関する重要な知見を提
供する。
原虫感染、ヒトア
ナプラズマ症、バ Parasitol Int.
32
ベシア症、トリパ 97(2023)102791.
ノソーマ症
ウシの生産はキルギスの国民経済に大きく貢献している。キルギスのウシのほとんどは、広域シス
テムで管理され、共同牧草地で放牧されている。その結果、外部寄生虫が蔓延しており、ウシに媒
介される様々な伝染病が多発している可能性がある。しかし、ウシに感染する媒介性病原体(VBP)
の疫学が依然として不明なため、キルギスではそのような感染症を防除する方法を利用できない。
そこで本研究では、キルギスのウシのVBPを調査することを目的とした。キルギスのウシ319頭から
血液DNAサンプルを調製し、Babesia bovis 、Babesia bigemina 、Babesia naoakii 、Theileria
annulata 、Theileria orientalis 、Trypanosoma evansi 、Trypanosoma theileri 、Anaplasma marginale
感染を検出するための特異的PCRアッセイでスクリーニングした。その結果、調査対象のウシは8つ
の病原体のうち6つに感染しており、B. naoakii とTry. evansi は除外された。最も多かった病原体はT.
orientalis (84.3%)で、次いでB. bigemina (47.6%)、T. annulata (16.6%)、A. marginale (11.6%)、
Try. theileri (7.2%)、B. bovis (2.5%)であった。Babesia 属特異的18S rRNA PCRを用いたB. bovis
及びB. bigemina 陰性検体の追加スクリーニングにより2検体で陽性が特定され、塩基配列解析から
それぞれBabesia major 又はBabesia occultans に感染していることが確認された。我々の知る限り、
これはキルギスのウシにおけるB. bovis 、B. bigemina 、B. occultans 、Try. theileri 及びA. marginale
感染の最初の報告である。この結果よりキルギスのウシはVBPによる感染症のリスクが高いことが
示唆された。
29
30
31
7 / 13
感染症(PT)
出典
概要
ロタウイルス感
染
従来とは異なるG3P[6]遺伝子型を有するヒトロタウイルス株は、世界各地の下痢症患者から散発的
に検出されている。しかし、全ゲノムの塩基配列が決定され、特徴が明らかにされているアジア諸国
(中国、インドネシア、ベトナム)由来のヒトG3P[6]株は3株のみであることから、アジアにおける
G3P[6]株の正確な起源と進化は未だ解明されていない。本報告では、日本(千葉)において、2020
年12月に急性胃腸炎で入院した3カ月齢の男児の便検体から検出されたG3P[6]株(RVA/Humanwt/JPN/SO1199/2020/G3P[6])の全ゲノムの配列を決定し、特徴付けを行った。完全ゲノム解析
で、SO1199株は独特のWa様遺伝子群配列:G3-P[6]-I5-R1-C1-M1-A8-N1-T1-E1-H1を有するこ
とが明らかになった。VP6遺伝子型I5及びNSP1遺伝子型A8は、ブタロタウイルス株において一般的
に認められる。さらに、系統解析により、SO1199株の11遺伝子はすべて、ブタ又はブタ様ヒトロタウ
Infection, Genetics and
イルスの遺伝子と密接に関連しており、したがってブタ由来であることが示唆された。このように、
Evolution.
SO1199株はブタ様遺伝子のバックボーンを保有していることが示されたことから、ブタロタウイルス
115(2023)105507
株の種間伝播の結果である可能性が高い。SO1199株の全11遺伝子が、これまでに同定されたアジ
アを含む世界中のブタ様ヒトG3P[6]株とは異なるクラスター内に系統発生学的に位置しており、ブタ
からヒトへの独立した人畜共通感染症の発生が示唆された。SO1199株がブタ由来であることは、ヒト
とブタなどの家畜との間に近接することによる人獣共通感染症の発生を示唆している。実際、
SO1199株が検出された千葉県はブタが多く、ブタとヒトが密接に接触している一大畜産地である。し
たがって、SO1199株が検出された男児は直接的あるいは間接的にブタと接触していた可能性があ
る。しかし、SO1199株の遺伝子について、ブタ株よりブタ様ヒト株の方が配列同一性が高く、SO1199
株の進化経路はブタからの直接的な種間伝播よりも複雑かもしれず、ヒトと動物の両方でロタウイル
ス株を調査する必要がある。
ロタウイルス感
染
本研究では、ケニアの下痢患児(生後48カ月の女児)の便検体から検出されたG4P[6]株(RVA
(Group A rotavirus)/Human-wt/KEN/KCH148/2019/G4P[6])の全ゲノムの特性を評価した。完全
ゲノム解析により、KCH148株はWa様遺伝子群配列:G4-P[6]-I1-R1-C1-M1-A1-N1-T7-E1-H1を
有することが判明した。NSP3遺伝子型T7は、ブタロタウイルス株において一般的に認められ、系統
Infection, Genetics and 解析の結果、KCH148株の11遺伝子のうち10遺伝子がブタ由来であり、残りのNSP2遺伝子はヒト由
Evolution.
来であることが示された。KCH148株はブタロタウイルスの骨格を有しており、ブタ由来である可能性
96(2021)105133
が高いことが判明した。また、KCH148株がブタ由来であることは、ヒトと家畜が近接していることに起
因する人獣共通感染も示唆しており、特にヒトとブタなどの家畜との密接な接触があるアフリカの発
展途上国で顕著である。実際、KCH148株が発見されたKiambu郡は、ヒトと家畜が密接に接触してい
る農村部を有する都市周辺の郡であり、KCH148株が検出された患児がブタと接触していた可能性
がある。
ロタウイルス感
染
Virus Genes.
57(2021)338-357
レオウイルス科に属するA群ロタウイルス(RVA)は、ヒト及び世界中の多くの動物種の双方において
重度の胃腸炎の重要な病原体である。ほとんどのRVAは宿主に限定されているようであるが、再集
合を伴う又は伴わないRVAの種間伝播が観察されている。ヒトRVAsに関しては、6G(G1~G4、G9、
G12)と3P( P[4]、P[6]、P[8])が主要な遺伝子型と考えられているが、ブタRVAsはほとんどが5G(G3
~G5、G9、G11)と2P(P[6]、P[7])の遺伝子型に割り当てられている。ヒトではG4は通常P[8]遺伝子
型と関連しているが、ブタではP[6]と結合したG4が主に検出される。一方、世界各地の下痢症患者
では従来とは異なるG4P[6]遺伝子型が散発的に検出されている。これらのG4P[6]株のいくつかの全
ゲノムの塩基配列が決定され、特徴が明らかにされたことから、これらのヒト株の大部分がブタ由来
であることを示す証拠が得られた。しかし、ヒトG4P[6]株の正確な進化パターンは、独特で菌株特異
的な遺伝子型群を保有しているため、未だ解明されていない。本研究では、2014年と2015年に、タイ
の重症下痢症入院患者において、2つのG4P[6]株、DU2014-259とPK2015-1-0001を同定し、計514
のRVA陽性糞便検体をG/P遺伝子型タイピングにより検査した。結果、2株ともG4-P[6]-I1-R1-C1M1-A8-N1-T1-E1-H1という独特のWa様遺伝子型群を示した。NSP1遺伝子型A8はブタロタウイル
ス株で一般的に見られる遺伝子型である。さらに、系統解析の結果、DU2014-259株及びPK2015-10001株の11遺伝子はいずれもブタ由来と考えられた。一方、11遺伝子セグメントのうち9遺伝子セグ
メント(VP4、VP6、VP1~VP3、NSP2~NSP5)において一貫して異なるクラスターを形成しており、ブ
タからヒトへの独立した種間伝播が起こっていることが強く示唆された。この結果は、人獣共通感染
症であるG4P[6]株の起源や、ブタとヒトのロタウイルス株の動的相互作用に関する重要な知見を提
供する。
原虫感染、ヒトア
ナプラズマ症、バ Parasitol Int.
32
ベシア症、トリパ 97(2023)102791.
ノソーマ症
ウシの生産はキルギスの国民経済に大きく貢献している。キルギスのウシのほとんどは、広域シス
テムで管理され、共同牧草地で放牧されている。その結果、外部寄生虫が蔓延しており、ウシに媒
介される様々な伝染病が多発している可能性がある。しかし、ウシに感染する媒介性病原体(VBP)
の疫学が依然として不明なため、キルギスではそのような感染症を防除する方法を利用できない。
そこで本研究では、キルギスのウシのVBPを調査することを目的とした。キルギスのウシ319頭から
血液DNAサンプルを調製し、Babesia bovis 、Babesia bigemina 、Babesia naoakii 、Theileria
annulata 、Theileria orientalis 、Trypanosoma evansi 、Trypanosoma theileri 、Anaplasma marginale
感染を検出するための特異的PCRアッセイでスクリーニングした。その結果、調査対象のウシは8つ
の病原体のうち6つに感染しており、B. naoakii とTry. evansi は除外された。最も多かった病原体はT.
orientalis (84.3%)で、次いでB. bigemina (47.6%)、T. annulata (16.6%)、A. marginale (11.6%)、
Try. theileri (7.2%)、B. bovis (2.5%)であった。Babesia 属特異的18S rRNA PCRを用いたB. bovis
及びB. bigemina 陰性検体の追加スクリーニングにより2検体で陽性が特定され、塩基配列解析から
それぞれBabesia major 又はBabesia occultans に感染していることが確認された。我々の知る限り、
これはキルギスのウシにおけるB. bovis 、B. bigemina 、B. occultans 、Try. theileri 及びA. marginale
感染の最初の報告である。この結果よりキルギスのウシはVBPによる感染症のリスクが高いことが
示唆された。
29
30
31
7 / 13