











よむ、つかう、まなぶ。
【資料No.2】塩野義製薬株式会社提出資料 (3 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_26901.html |
| 出典情報 | 薬事・食品衛生審議会 薬事分科会(令和4年度第3回 7/20)、医薬品第二部会(令和4年度第6回 7/20)(合同開催)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
2. 本剤の有効性
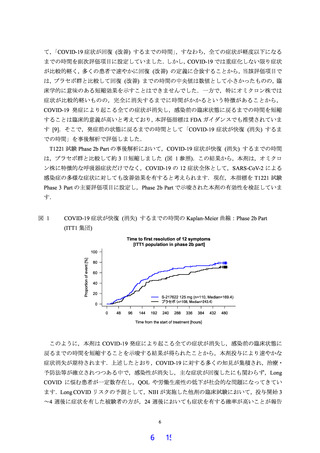
本剤の緊急承認申請にあたり,T1221 試験の Phase 2a Part 及び Phase 2b Part の結果に基づき,
本剤の有効性を評価しました.Phase 2a Part では抗ウイルス効果を探索的に確認することを,
Phase 2b Part では早期の臨床症状改善効果及び抗ウイルス効果を確認することを目的としまし
た.
軽症/中等症及び無症候の SARS-CoV-2 感染者を対象とした Phase 2a Part において,主要評価
項目として設定したウイルス力価のベースライン (投与前) からの変化量,及び副次評価項目
として設定したウイルス RNA 量のベースラインからの変化量等の結果から,本剤の抗ウイル
ス効果が確認されました.また,臨床症状に対しては,デルタ株以前の流行株で SARS-CoV-2 感
染者に特徴的な症状として報告されていた 12 症状 (けん怠感 [疲労感],筋肉痛又は体の痛み,
頭痛,悪寒/発汗,熱っぽさ又は発熱,鼻水又は鼻づまり,喉の痛み,咳,息切れ [呼吸困難],
吐き気,嘔吐,下痢) の合計スコアの低下が認められたことから,臨床症状の改善効果について
も期待できる結果が得られました.
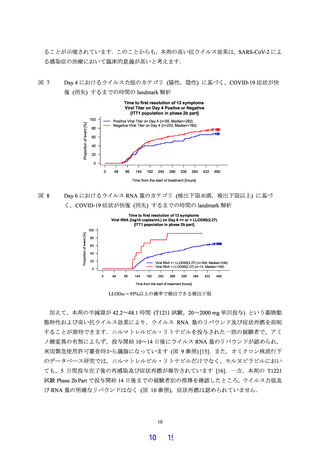
続いて実施した軽症/中等症の SARS-CoV-2 感染者を対象とした Phase 2b Part では,Phase 2a
Part の結果に基づき,抗ウイルス効果に関する評価項目として Day 4 (治験薬投与開始 3 日後)
における SARS-CoV-2 のウイルス力価のベースラインからの変化量と,臨床症状改善効果の評
価項目として COVID-19 の 12 症状合計スコアの Day 1 (治験薬投与開始日) から Day 6 (治験薬
投与開始 5 日後) までの単位時間あたりの変化量を設定し,これら 2 つを主要評価項目
(Co-primary endpoint) としました.Co-primary endpoint であるため,2 つの評価項目の両方でプ
ラセボ群に対する有意な差が認められた場合に限り,本試験の成功基準を満たしたと規定しま
した.Day 4 における SARS-CoV-2 のウイルス力価のベースラインからの変化量では,プラセボ
群と比較して有意な減少が認められましたが,COVID-19 の 12 症状合計スコアの Day 1 から
Day 6 までの単位時間あたりの変化量では,プラセボ群と比較して有意差は認められず,事前に
規定した本試験の成功基準は満たしませんでした.
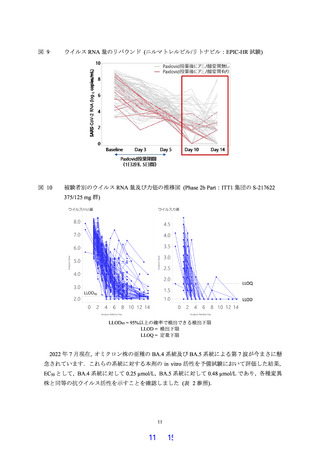
本試験の成功基準を満たさなかった理由として,Phase 2b Part 実施時の流行株であるオミク
ロン株は,デルタ株以前の流行株と比較して感染時の臨床的な特徴が大きく異なっていたこと
が影響したと考えます.具体的には,オミクロン株の感染者では,呼吸器や発熱以外の症状が
ほとんど認められない又はその程度は軽いことから,デルタ株以前の株の感染者に特徴的な症
状として報告されていた 12 症状を対象とした合計スコアの変化量では,臨床症状の改善効果を
捉えることは難しく,統計学的な有意差を確認することができなかったと考えます.
背景にて上述したとおり,SARS-CoV-2 による感染症に対する治療薬の開発においては,流行
株の遷移やワクチン接種状況など,臨床試験の評価対象となる集団や公衆衛生上の課題が変わ
りゆく中で,抗ウイルス薬としての有効性を確認・検証していくことが求められています.そ
の中で,臨床試験での評価指標についてはこれまで様々な議論が続けられており,T1221 試験
の開始時点に確固たる評価指標が定まっていなかったことから,本試験では本剤の有効性を多
角的に評価するために,上述の主要評価項目以外にも,様々な評価項目を副次評価項目として
事前に規定し,さらには,試験開始後に明らかになってきた臨床上やウイルス学的な特徴を踏
まえて,事後解析を実施しました.その結果を以下に示します (表 1 参照).
3
3 / 15
本剤の緊急承認申請にあたり,T1221 試験の Phase 2a Part 及び Phase 2b Part の結果に基づき,
本剤の有効性を評価しました.Phase 2a Part では抗ウイルス効果を探索的に確認することを,
Phase 2b Part では早期の臨床症状改善効果及び抗ウイルス効果を確認することを目的としまし
た.
軽症/中等症及び無症候の SARS-CoV-2 感染者を対象とした Phase 2a Part において,主要評価
項目として設定したウイルス力価のベースライン (投与前) からの変化量,及び副次評価項目
として設定したウイルス RNA 量のベースラインからの変化量等の結果から,本剤の抗ウイル
ス効果が確認されました.また,臨床症状に対しては,デルタ株以前の流行株で SARS-CoV-2 感
染者に特徴的な症状として報告されていた 12 症状 (けん怠感 [疲労感],筋肉痛又は体の痛み,
頭痛,悪寒/発汗,熱っぽさ又は発熱,鼻水又は鼻づまり,喉の痛み,咳,息切れ [呼吸困難],
吐き気,嘔吐,下痢) の合計スコアの低下が認められたことから,臨床症状の改善効果について
も期待できる結果が得られました.
続いて実施した軽症/中等症の SARS-CoV-2 感染者を対象とした Phase 2b Part では,Phase 2a
Part の結果に基づき,抗ウイルス効果に関する評価項目として Day 4 (治験薬投与開始 3 日後)
における SARS-CoV-2 のウイルス力価のベースラインからの変化量と,臨床症状改善効果の評
価項目として COVID-19 の 12 症状合計スコアの Day 1 (治験薬投与開始日) から Day 6 (治験薬
投与開始 5 日後) までの単位時間あたりの変化量を設定し,これら 2 つを主要評価項目
(Co-primary endpoint) としました.Co-primary endpoint であるため,2 つの評価項目の両方でプ
ラセボ群に対する有意な差が認められた場合に限り,本試験の成功基準を満たしたと規定しま
した.Day 4 における SARS-CoV-2 のウイルス力価のベースラインからの変化量では,プラセボ
群と比較して有意な減少が認められましたが,COVID-19 の 12 症状合計スコアの Day 1 から
Day 6 までの単位時間あたりの変化量では,プラセボ群と比較して有意差は認められず,事前に
規定した本試験の成功基準は満たしませんでした.
本試験の成功基準を満たさなかった理由として,Phase 2b Part 実施時の流行株であるオミク
ロン株は,デルタ株以前の流行株と比較して感染時の臨床的な特徴が大きく異なっていたこと
が影響したと考えます.具体的には,オミクロン株の感染者では,呼吸器や発熱以外の症状が
ほとんど認められない又はその程度は軽いことから,デルタ株以前の株の感染者に特徴的な症
状として報告されていた 12 症状を対象とした合計スコアの変化量では,臨床症状の改善効果を
捉えることは難しく,統計学的な有意差を確認することができなかったと考えます.
背景にて上述したとおり,SARS-CoV-2 による感染症に対する治療薬の開発においては,流行
株の遷移やワクチン接種状況など,臨床試験の評価対象となる集団や公衆衛生上の課題が変わ
りゆく中で,抗ウイルス薬としての有効性を確認・検証していくことが求められています.そ
の中で,臨床試験での評価指標についてはこれまで様々な議論が続けられており,T1221 試験
の開始時点に確固たる評価指標が定まっていなかったことから,本試験では本剤の有効性を多
角的に評価するために,上述の主要評価項目以外にも,様々な評価項目を副次評価項目として
事前に規定し,さらには,試験開始後に明らかになってきた臨床上やウイルス学的な特徴を踏
まえて,事後解析を実施しました.その結果を以下に示します (表 1 参照).
3
3 / 15