



























よむ、つかう、まなぶ。
資料3-3 ストラテラカプセル及びストラテラ内用液にて検出された新規ニトロソアミンの限度値について(企業見解)[7.8MB] (15 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_42464.html |
| 出典情報 | 薬事審議会 医薬品等安全対策部会安全対策調査会(令和6年度第5回 8/28)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
Chemical Research in Toxicology
pubs.acs.org/crt
reactivity of noncarcinogens with carcinogenic nitrosamines
assuming that all the compounds undergo α-hydroxylation. We
reiterate the fact that the −COOH group is considered to be
only a substituent to understand the electron-withdrawing
effect. The AI values range from 70 ng/day (1) to 2520 ng/day
(2)19 which covers five categories described to predict AI
through the CPCA approach from the regulatory (in the range
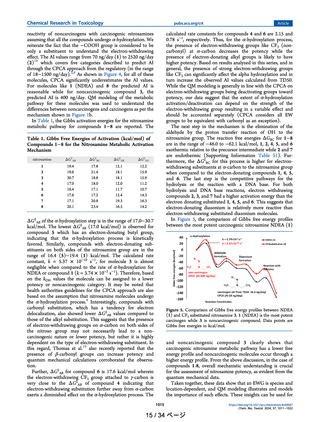
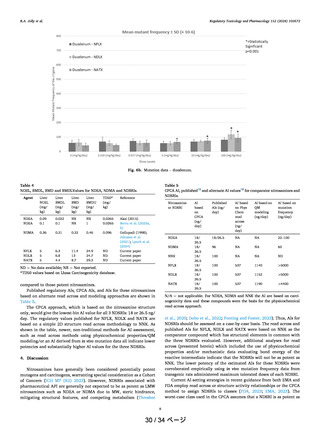
of 18−1500 ng/day).2,9 As shown in Figure 4, for all of these
molecules, CPCA significantly underestimates the AI values.
For molecules like 1 (NDEA) and 8 the predicted AI is
reasonable while for noncarcinogenic compound 3, the
predicted AI is 400 ng/day. QM modeling of the metabolic
pathway for these molecules was used to understand the
differences between noncarcinogens and carcinogens as per the
mechanism shown in Figure 1b.
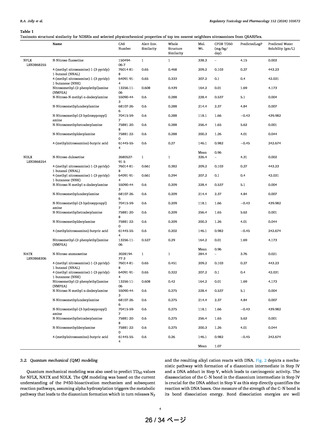
In Table 1, the Gibbs activation energies for the nitrosamine
metabolic pathway for compounds 1−8 are reported. The
calculated rate constants for compounds 4 and 6 are 2.15 and
0.78 s−1, respectively. Thus, for the α-hydroxylation process,
the presence of electron-withdrawing groups like CF3 (noncarbonyl) at α-carbon decreases the potency while the
presence of electron-donating alkyl groups is likely to have
higher potency. Based on results analyzed in this series, and in
general, the presence of strong electron-withdrawing groups
like CF3 can significantly affect the alpha hydroxylation and in
turn increase the observed AI values calculated from TD50.
While the QM modeling is generally in line with the CPCA on
electron-withdrawing groups being deactivating groups toward
potency, our data suggest that the extent of α-hydroxylation
activation/deactivation can depend on the strength of the
electron-withdrawing group resulting in a variable effect and
should be accounted separately (CPCA considers all EW
groups to be equivalent with carbonyl as an exception).
The next step in the mechanism is the elimination of the
aldehyde by the proton transfer reaction of OH to the
nitrosamine group. The reaction free energies ΔGBC for 1−8
are in the range of −46.0 to −62.1 kcal/mol, 1, 2, 4, 5, and 6
exothermic relative to the precursor intermediate while 2 and 7
are endothermic (Supporting Information Table S1). Furthermore, the ΔG‡BC for this process is higher for electronwithdrawing substituents at α-carbon to the nitrosamine group
when compared to the electron-donating compounds 1, 4, 5,
and 6. The last step is the competitive pathways for the
hydrolysis or the reaction with a DNA base. For both
hydrolysis and DNA base reactions, electron withdrawing
compounds 2, 3, and 7 had a higher activation energy than the
electron donating substituted 1, 4, 5, and 6. This suggests that
electron-donating diazonium is relatively more reactive than
electron-withdrawing substituted diazonium molecules.
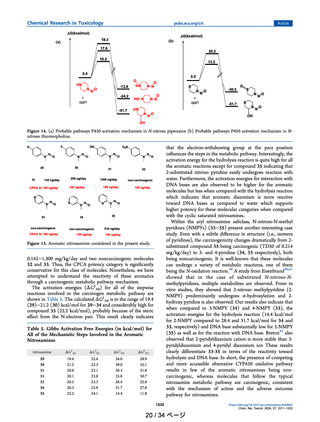
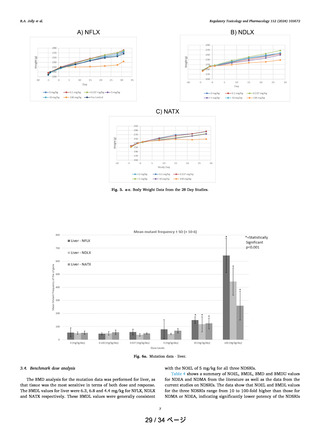
In Figure 5, the comparison of Gibbs free energy profiles
between the most potent carcinogenic nitrosamine NDEA (1)
Table 1. Gibbs Free Energies of Activation (kcal/mol) of
Compounds 1−8 for the Nitrosamine Metabolic Activation
Mechanism
nitrosamine
ΔG‡AB
ΔG‡BC
ΔG‡DE
ΔG‡DG
1
2
3
4
5
6
7
8
19.4
19.6
30.7
17.0
16.4
17.6
17.1
20.1
17.8
21.4
18.8
18.0
17.1
17.2
20.8
23.4
12.1
18.1
18.1
12.0
11.7
12.4
19.3
16.3
12.2
15.9
15.9
11.2
10.7
14.3
16.3
14.2
Article
ΔG‡AB of the α-hydroxylation step is in the range of 17.0−30.7
kcal/mol. The lowest ΔG‡AB (17.0 kcal/mol) is observed for
compound 5 which has an electron-donating butyl group,
indicating that the α-hydroxylation process is kinetically
favored. Similarly, compounds with electron-donating substituents on both sides of the nitrosamine group are in the
range of 16.4 (5)−19.4 (1) kcal/mol. The calculated rate
constant, k = 5.37 × 10−10 s−1, for molecule 3 is almost
negligible when compared to the rate of α-hydroxylation for
NDEA or compound 1 (k = 3.74 × 10−2 s−1). Therefore, based
on the kOH values the molecule can be assigned to a lower
potency or noncarcinogenic category. It may be noted that
health authorities guidelines for the CPCA approach are also
based on the assumption that nitrosamine molecules undergo
the α-hydroxylation process.9 Interestingly, compounds with
carbonyl substitution, which has a tendency for electron
delocalization, also showed lower ΔG‡AB values compared to
those of the alkyl substitution. This suggests that the presence
of electron-withdrawing groups on α-carbon on both sides of
the nitroso group may not necessarily lead to a noncarcinogenic nature or lower potency, but rather it is highly
dependent on the type of electron-withdrawing substituent. In
this regard, Thomas et al.19 also recently reported that the
presence of β-carbonyl groups can increase potency and
quantum mechanical calculations corroborated the observation.
Further, ΔG‡AB for compound 6 is 17.6 kcal/mol wherein
the electron-withdrawing CF3 group attached to γ-carbon is
very close to the ΔG‡AB of compound 4 indicating that
electron-withdrawing substitution further away from α-carbon
exerts a diminished effect on the α-hydroxylation process. The
Figure 5. Comparison of Gibbs free energy profiles between NDEA
(1) and CF3 substituted nitrosamine 3. 1 (NDEA) is the most potent
carcinogen while 3 is noncarcinogenic compound. Data points are
Gibbs free energies in kcal/mol.
and noncarcinogenic compound 3 clearly shows that
carcinogenic nitrosamine metabolic pathway has a lower free
energy profile and noncarcinogenic molecules occur through a
higher energy profile. From the above discussion, in the case of
compounds 1-8, overall mechanistic understanding is crucial
for the assessment of nitrosamine potency, as evident from the
quantum mechanical data.
Taken together, these data show that an EWG is species and
location-dependent, and QM modeling illustrates and models
the importance of such effects. These insights can be used for
1015
15 / 34 ページ
https://doi.org/10.1021/acs.chemrestox.4c00087
Chem. Res. Toxicol. 2024, 37, 1011−1022
pubs.acs.org/crt
reactivity of noncarcinogens with carcinogenic nitrosamines
assuming that all the compounds undergo α-hydroxylation. We
reiterate the fact that the −COOH group is considered to be
only a substituent to understand the electron-withdrawing
effect. The AI values range from 70 ng/day (1) to 2520 ng/day
(2)19 which covers five categories described to predict AI
through the CPCA approach from the regulatory (in the range
of 18−1500 ng/day).2,9 As shown in Figure 4, for all of these
molecules, CPCA significantly underestimates the AI values.
For molecules like 1 (NDEA) and 8 the predicted AI is
reasonable while for noncarcinogenic compound 3, the
predicted AI is 400 ng/day. QM modeling of the metabolic
pathway for these molecules was used to understand the
differences between noncarcinogens and carcinogens as per the
mechanism shown in Figure 1b.
In Table 1, the Gibbs activation energies for the nitrosamine
metabolic pathway for compounds 1−8 are reported. The
calculated rate constants for compounds 4 and 6 are 2.15 and
0.78 s−1, respectively. Thus, for the α-hydroxylation process,
the presence of electron-withdrawing groups like CF3 (noncarbonyl) at α-carbon decreases the potency while the
presence of electron-donating alkyl groups is likely to have
higher potency. Based on results analyzed in this series, and in
general, the presence of strong electron-withdrawing groups
like CF3 can significantly affect the alpha hydroxylation and in
turn increase the observed AI values calculated from TD50.
While the QM modeling is generally in line with the CPCA on
electron-withdrawing groups being deactivating groups toward
potency, our data suggest that the extent of α-hydroxylation
activation/deactivation can depend on the strength of the
electron-withdrawing group resulting in a variable effect and
should be accounted separately (CPCA considers all EW
groups to be equivalent with carbonyl as an exception).
The next step in the mechanism is the elimination of the
aldehyde by the proton transfer reaction of OH to the
nitrosamine group. The reaction free energies ΔGBC for 1−8
are in the range of −46.0 to −62.1 kcal/mol, 1, 2, 4, 5, and 6
exothermic relative to the precursor intermediate while 2 and 7
are endothermic (Supporting Information Table S1). Furthermore, the ΔG‡BC for this process is higher for electronwithdrawing substituents at α-carbon to the nitrosamine group
when compared to the electron-donating compounds 1, 4, 5,
and 6. The last step is the competitive pathways for the
hydrolysis or the reaction with a DNA base. For both
hydrolysis and DNA base reactions, electron withdrawing
compounds 2, 3, and 7 had a higher activation energy than the
electron donating substituted 1, 4, 5, and 6. This suggests that
electron-donating diazonium is relatively more reactive than
electron-withdrawing substituted diazonium molecules.
In Figure 5, the comparison of Gibbs free energy profiles
between the most potent carcinogenic nitrosamine NDEA (1)
Table 1. Gibbs Free Energies of Activation (kcal/mol) of
Compounds 1−8 for the Nitrosamine Metabolic Activation
Mechanism
nitrosamine
ΔG‡AB
ΔG‡BC
ΔG‡DE
ΔG‡DG
1
2
3
4
5
6
7
8
19.4
19.6
30.7
17.0
16.4
17.6
17.1
20.1
17.8
21.4
18.8
18.0
17.1
17.2
20.8
23.4
12.1
18.1
18.1
12.0
11.7
12.4
19.3
16.3
12.2
15.9
15.9
11.2
10.7
14.3
16.3
14.2
Article
ΔG‡AB of the α-hydroxylation step is in the range of 17.0−30.7
kcal/mol. The lowest ΔG‡AB (17.0 kcal/mol) is observed for
compound 5 which has an electron-donating butyl group,
indicating that the α-hydroxylation process is kinetically
favored. Similarly, compounds with electron-donating substituents on both sides of the nitrosamine group are in the
range of 16.4 (5)−19.4 (1) kcal/mol. The calculated rate
constant, k = 5.37 × 10−10 s−1, for molecule 3 is almost
negligible when compared to the rate of α-hydroxylation for
NDEA or compound 1 (k = 3.74 × 10−2 s−1). Therefore, based
on the kOH values the molecule can be assigned to a lower
potency or noncarcinogenic category. It may be noted that
health authorities guidelines for the CPCA approach are also
based on the assumption that nitrosamine molecules undergo
the α-hydroxylation process.9 Interestingly, compounds with
carbonyl substitution, which has a tendency for electron
delocalization, also showed lower ΔG‡AB values compared to
those of the alkyl substitution. This suggests that the presence
of electron-withdrawing groups on α-carbon on both sides of
the nitroso group may not necessarily lead to a noncarcinogenic nature or lower potency, but rather it is highly
dependent on the type of electron-withdrawing substituent. In
this regard, Thomas et al.19 also recently reported that the
presence of β-carbonyl groups can increase potency and
quantum mechanical calculations corroborated the observation.
Further, ΔG‡AB for compound 6 is 17.6 kcal/mol wherein
the electron-withdrawing CF3 group attached to γ-carbon is
very close to the ΔG‡AB of compound 4 indicating that
electron-withdrawing substitution further away from α-carbon
exerts a diminished effect on the α-hydroxylation process. The
Figure 5. Comparison of Gibbs free energy profiles between NDEA
(1) and CF3 substituted nitrosamine 3. 1 (NDEA) is the most potent
carcinogen while 3 is noncarcinogenic compound. Data points are
Gibbs free energies in kcal/mol.
and noncarcinogenic compound 3 clearly shows that
carcinogenic nitrosamine metabolic pathway has a lower free
energy profile and noncarcinogenic molecules occur through a
higher energy profile. From the above discussion, in the case of
compounds 1-8, overall mechanistic understanding is crucial
for the assessment of nitrosamine potency, as evident from the
quantum mechanical data.
Taken together, these data show that an EWG is species and
location-dependent, and QM modeling illustrates and models
the importance of such effects. These insights can be used for
1015
15 / 34 ページ
https://doi.org/10.1021/acs.chemrestox.4c00087
Chem. Res. Toxicol. 2024, 37, 1011−1022