























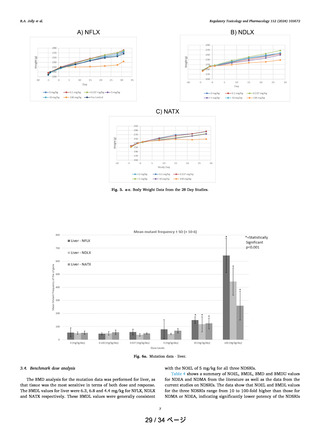
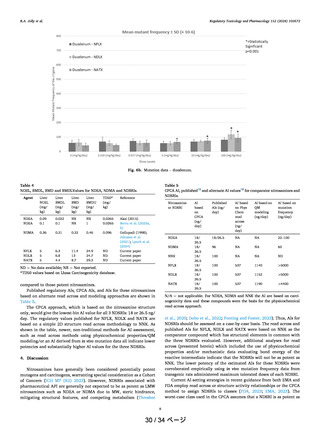




よむ、つかう、まなぶ。
資料3-3 ストラテラカプセル及びストラテラ内用液にて検出された新規ニトロソアミンの限度値について(企業見解)[7.8MB] (17 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_42464.html |
| 出典情報 | 薬事審議会 医薬品等安全対策部会安全対策調査会(令和6年度第5回 8/28)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
Chemical Research in Toxicology
pubs.acs.org/crt
et al.19 reported that β-carbonyl substitution increases the
potency. The QM calculations illustrate that α-hydroxylation is
easier if the nitrosamine has a β-carbonyl substitution.
The CPCA-derived potency score for the molecules 9−19 is
simply the α-hydrogen score, which is 1 (3 hydrogens on one
side and 2 hydrogens on the other) and falls under the
category 1 corresponding to 18 ng/day. Molecules 11, 12, 16,
and 19 all have a β-hydroxy group as the deactivating structural
feature with a potency score of 2 and therefore fall under
category 2 with a 100 ng/day AI limit. For these molecules, the
CPCA category mostly underestimates the AI range calculated
from the actual TD50. However, molecule 17 falls under
category 4 corresponding to a 1500 ng/day AI limit (actual AI
is 982 ng/day). This is an example where CPCA overestimates
the potency.
The molecule 15 has a β-substituted N(CH3)2 group with
AI of 3830 ng/day, and CPCA predicted AI is 18 ng/day. For
this molecule, possible alternate hydroxylation mechanisms via
dealkylation or oxygenation were also considered and are
shown in Figure 7. The calculated activation energy for the N-
Article
of observed α-hydroxylation (ΔG‡AB′ > ΔG‡AB), we performed
transition state structure calculations (TSBC) for the proton
transfer mechanism on both sides of α-hydroxylated molecules.
The aldehyde formation step is endergonic relative to the
hydroxylation step for the ΔGBC when compared with ΔGBC′
(Supporting Information). The activation energies for the
aldehyde formation step follow a similar trend ΔG‡BC >
ΔG‡BC′ which further confirms that alkyl aldehyde formation is
preferred both kinetically as well as thermodynamically (Table
S1). Wenzel et al.20 also found a similar observation that
methyl group elimination had a high activation energy and
endergonic in the case of N-methyl, long-chain nitrosamines.
Therefore, from the above discussion, for all molecules 9−19,
the methyl diazonium intermediate or the corresponding alkyl
diazonium intermediate can be formed. The activation energies
for the hydrolysis and DNA base are reported in Table 2.
Overall, the quantum mechanical data provide a further
reactivity-based explanation to the experimentally observed
potency trends of N-methyl nitrosamines. This further suggests
that the CPCA scoring and classification can be augmented
with QM analysis to understand and give a better AI
estimation for NDSRIs.
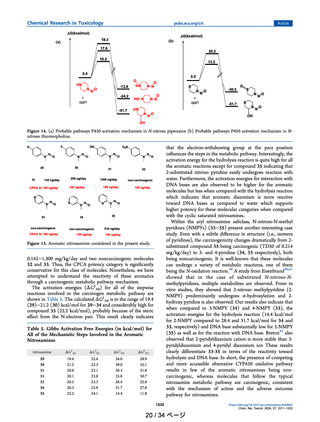
Ring Nitrosamines. N-Nitroso Pyrrolidines. Substituted
N-nitroso pyrrolidine compounds (20−24) are shown in
Figure 8. In this class of molecules, a nitroso group is attached
Figure 8. N-nitroso pyrrolidine compounds. AI from TD50 and
CPCA predicted AI limits is also shown.
to an amine nitrogen which is a part of a five-membered ring.
These compounds represent the effect of OH substitution
(21), the presence of heteroatom which is part of the ring
(22), electron-withdrawing COOH (23), and aromatic
substitution, NNN (24). In the CPCA, this class of molecules
has a deactivating feature score of +3 which would be expected
to have low potency because of the high CPCA score. The
TD50 values for these molecules range from 0.0957 to 7.65
mg/kg/day. The highest potency is observed for a m-pyridine
substituted at the α-position N-nitroso pyrrolidone (NNN, 24;
0.0957 mg/kg/day). For 20−24, we evaluated the reaction
energy profiles for all the metabolic steps involved in the
carcinogenic metabolic pathway shown in Figure 1a. The
ΔG‡AB is in a narrow range of 18.4−20.3 kcal/mol (rate
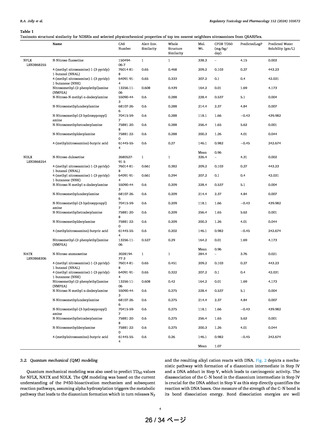
constants are in the range of 2.4 × 10−1−8.2 × 10−3 s−1). The
relative rate constants of α-hydroxylation with respect to
compound 20 are 0.18, 4.57, 4.57, and 2.75-fold. Figure 9
shows the optimized transition state geometries for compounds 20−24. The geometrical data are very similar in
compounds 20−24 and the C−H bond distances are in the
range of 1.307−1.323 Å and the O−H bond distances are in
the range of 1.217−1.240 Å showing no significant changes
(Table 3). These data indicate that probably the other steps in
this metabolic pathway could be playing a rate-limiting role in
such variation as discussed below.
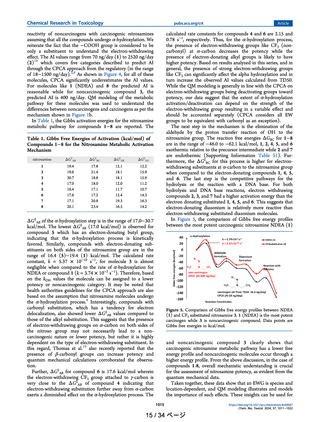
Figure 7. Probable competitive pathways α-hydroxylation, Ndealkylation and N-oxygenation of 15 by cpd1. Gibbs activation
free energies represent that α-hydroxylation is not the preferred
pathway, despite having two CH2 groups at the α-position.
dealkylation is 7.4 kcal/mol and for N-oxygenation is 12.9
kcal/mol, which is substantially lower than α-hydroxylation
(19.0 kcal/mol).35 It indicates that compound 15 can undergo
other competitive metabolic mechanisms, and hence, the
carcinogenic mechanism is kinetically least preferred. Thus, the
variation observed in the AI also can strongly depend on the
potential of undergoing alternate metabolism, and that needs
to be carefully considered while assessing the AI limits for
NDSRIs. Overall, the discussion above further shows that the
QM calculations can be used as a viable tool to further evaluate
and adjust the AI limits for NDSRI compounds where CPCA
falls short.
The AI values for compounds 9, 10, 12, 13, and 18 are 96,
50, 46, 17, and 10 ng/day, respectively which supports the fact
that nitrosamines with N-methyl and N-alkyl combination with
α−C-H in the alkyl group have low AI when compared to
doubly substituted compounds such as 16 and 19 (AI is 646
and 95,200 ng/day, respectively). To further confirm the trend
1017
17 / 34 ページ
https://doi.org/10.1021/acs.chemrestox.4c00087
Chem. Res. Toxicol. 2024, 37, 1011−1022
pubs.acs.org/crt
et al.19 reported that β-carbonyl substitution increases the
potency. The QM calculations illustrate that α-hydroxylation is
easier if the nitrosamine has a β-carbonyl substitution.
The CPCA-derived potency score for the molecules 9−19 is
simply the α-hydrogen score, which is 1 (3 hydrogens on one
side and 2 hydrogens on the other) and falls under the
category 1 corresponding to 18 ng/day. Molecules 11, 12, 16,
and 19 all have a β-hydroxy group as the deactivating structural
feature with a potency score of 2 and therefore fall under
category 2 with a 100 ng/day AI limit. For these molecules, the
CPCA category mostly underestimates the AI range calculated
from the actual TD50. However, molecule 17 falls under
category 4 corresponding to a 1500 ng/day AI limit (actual AI
is 982 ng/day). This is an example where CPCA overestimates
the potency.
The molecule 15 has a β-substituted N(CH3)2 group with
AI of 3830 ng/day, and CPCA predicted AI is 18 ng/day. For
this molecule, possible alternate hydroxylation mechanisms via
dealkylation or oxygenation were also considered and are
shown in Figure 7. The calculated activation energy for the N-
Article
of observed α-hydroxylation (ΔG‡AB′ > ΔG‡AB), we performed
transition state structure calculations (TSBC) for the proton
transfer mechanism on both sides of α-hydroxylated molecules.
The aldehyde formation step is endergonic relative to the
hydroxylation step for the ΔGBC when compared with ΔGBC′
(Supporting Information). The activation energies for the
aldehyde formation step follow a similar trend ΔG‡BC >
ΔG‡BC′ which further confirms that alkyl aldehyde formation is
preferred both kinetically as well as thermodynamically (Table
S1). Wenzel et al.20 also found a similar observation that
methyl group elimination had a high activation energy and
endergonic in the case of N-methyl, long-chain nitrosamines.
Therefore, from the above discussion, for all molecules 9−19,
the methyl diazonium intermediate or the corresponding alkyl
diazonium intermediate can be formed. The activation energies
for the hydrolysis and DNA base are reported in Table 2.
Overall, the quantum mechanical data provide a further
reactivity-based explanation to the experimentally observed
potency trends of N-methyl nitrosamines. This further suggests
that the CPCA scoring and classification can be augmented
with QM analysis to understand and give a better AI
estimation for NDSRIs.
Ring Nitrosamines. N-Nitroso Pyrrolidines. Substituted
N-nitroso pyrrolidine compounds (20−24) are shown in
Figure 8. In this class of molecules, a nitroso group is attached
Figure 8. N-nitroso pyrrolidine compounds. AI from TD50 and
CPCA predicted AI limits is also shown.
to an amine nitrogen which is a part of a five-membered ring.
These compounds represent the effect of OH substitution
(21), the presence of heteroatom which is part of the ring
(22), electron-withdrawing COOH (23), and aromatic
substitution, NNN (24). In the CPCA, this class of molecules
has a deactivating feature score of +3 which would be expected
to have low potency because of the high CPCA score. The
TD50 values for these molecules range from 0.0957 to 7.65
mg/kg/day. The highest potency is observed for a m-pyridine
substituted at the α-position N-nitroso pyrrolidone (NNN, 24;
0.0957 mg/kg/day). For 20−24, we evaluated the reaction
energy profiles for all the metabolic steps involved in the
carcinogenic metabolic pathway shown in Figure 1a. The
ΔG‡AB is in a narrow range of 18.4−20.3 kcal/mol (rate
constants are in the range of 2.4 × 10−1−8.2 × 10−3 s−1). The
relative rate constants of α-hydroxylation with respect to
compound 20 are 0.18, 4.57, 4.57, and 2.75-fold. Figure 9
shows the optimized transition state geometries for compounds 20−24. The geometrical data are very similar in
compounds 20−24 and the C−H bond distances are in the
range of 1.307−1.323 Å and the O−H bond distances are in
the range of 1.217−1.240 Å showing no significant changes
(Table 3). These data indicate that probably the other steps in
this metabolic pathway could be playing a rate-limiting role in
such variation as discussed below.
Figure 7. Probable competitive pathways α-hydroxylation, Ndealkylation and N-oxygenation of 15 by cpd1. Gibbs activation
free energies represent that α-hydroxylation is not the preferred
pathway, despite having two CH2 groups at the α-position.
dealkylation is 7.4 kcal/mol and for N-oxygenation is 12.9
kcal/mol, which is substantially lower than α-hydroxylation
(19.0 kcal/mol).35 It indicates that compound 15 can undergo
other competitive metabolic mechanisms, and hence, the
carcinogenic mechanism is kinetically least preferred. Thus, the
variation observed in the AI also can strongly depend on the
potential of undergoing alternate metabolism, and that needs
to be carefully considered while assessing the AI limits for
NDSRIs. Overall, the discussion above further shows that the
QM calculations can be used as a viable tool to further evaluate
and adjust the AI limits for NDSRI compounds where CPCA
falls short.
The AI values for compounds 9, 10, 12, 13, and 18 are 96,
50, 46, 17, and 10 ng/day, respectively which supports the fact
that nitrosamines with N-methyl and N-alkyl combination with
α−C-H in the alkyl group have low AI when compared to
doubly substituted compounds such as 16 and 19 (AI is 646
and 95,200 ng/day, respectively). To further confirm the trend
1017
17 / 34 ページ
https://doi.org/10.1021/acs.chemrestox.4c00087
Chem. Res. Toxicol. 2024, 37, 1011−1022