
































よむ、つかう、まなぶ。
参考資料4:総薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会報告書 (30 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_43236.html |
| 出典情報 | 厚生科学審議会 臨床研究部会(第36回 9/4)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
(2)検討会における議論
事務局作成の資料19に基づいて議論が進められたが、その中では、日米欧の変更管
理の手続の概要(図9)や、実際の変更カテゴリの日本と欧米間の違いに関する製薬
協のアンケート結果などが示された。また、日本製薬工業協会・柏谷構成員からは、
製薬業界の意見20が発表された。
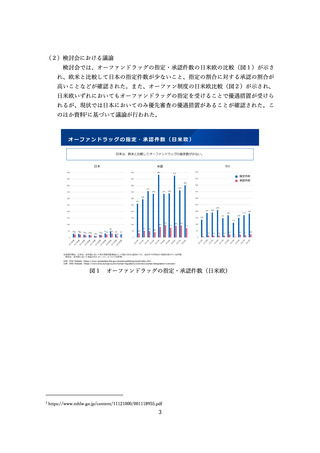
日米欧の変更管理の手続の概要
⚫ ICH Q12(医薬品のライフサイクルマネジメント)において、製造方法等の変更管理における薬事手続は3つにわけて例示されているが、
日本では薬事手続は2つのみ。また、年次報告の仕組みもない。
⚫ なお、以下の表は、ICH Q12の分類に従って3極の分類を当てはめたものであり、実際の変更事項の分類が3極で一致するものではない。
ICH Q12の分類
事前承認
届出・中リスク
米国
EU
日本
PAS(Prior Approval Supplement )
Type II Variation
一部変更承認申請
変更前に事前申請
変更前に事前申請
変更前に事前申請
CBE30
Type IB Variation
変更計画を提出し、受領連絡(提出から
14日以内)から30日以内に連絡がなけ
れば変更可
変更計画を提出し、受領連絡(提出から
7日程度)から30日以内に連絡がなけれ
ば変更可
中リスクに対応する
カテゴリがない
Type IAIN Variation
変更計画を提出し、受領連絡後に変更可
変更後、速やかに変更内容を提出。有効
or無効のフィードバックが30日以内にあ
る。
Annual Report
Type IA Variation
変更事項を1年に1回提出
変更後12か月以内に変更内容を提出。他
の変更と併せて、年次報告とすることも
可能。
CBE0
届出・低リスク
軽微変更届出
変更後30日以内に届出
※実際には欧米では届出相当の手続も日本
では軽微変更ではなく一部変更申請が求め
られる場合がある(次ページ以降参照)
Annual Report、Type IA Valiation
のような、年次報告の仕組みがない
報告不要
3
図9 日米欧の変更管理の手続の概要
(3)対応の方向性
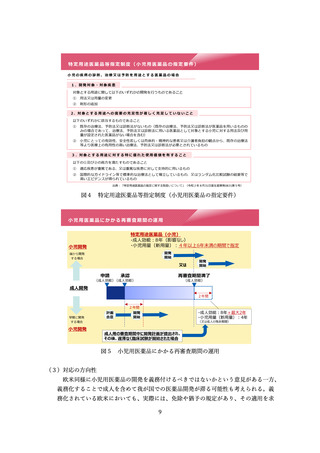
①
中等度変更事項の導入
医薬品の製造方法等の変更管理については、欧米と同様に、変更案を提出し、短
期間の確認期間を経て変更を行うことができる新たな変更カテゴリとして「中等度
変更事項」を導入すべきこととした。
制度の詳細やフィージビリティを検討するため、まずは対象を限定して試行的に
導入すべきこととした。
試行における「中等度変更事項」の対象については、変更内容のリスクの程度に
基づき、①初回承認申請又は一変申請の審査においてあらかじめ「中等度変更事項」
として特定された事項、及び②変更が生じた都度の PMDA 相談で中等度変更事項へ
の該当性を確認された事項とした。
また、試行においての「中等度変更事項」に係る薬事手続は、現行の一変申請の
一類型とした上で、その審査を短期間で実施することとした。
19
https://www.mhlw.go.jp/content/11121000/001155992.pdf
20
https://www.mhlw.go.jp/content/11121000/001155993.pdf
30
事務局作成の資料19に基づいて議論が進められたが、その中では、日米欧の変更管
理の手続の概要(図9)や、実際の変更カテゴリの日本と欧米間の違いに関する製薬
協のアンケート結果などが示された。また、日本製薬工業協会・柏谷構成員からは、
製薬業界の意見20が発表された。
日米欧の変更管理の手続の概要
⚫ ICH Q12(医薬品のライフサイクルマネジメント)において、製造方法等の変更管理における薬事手続は3つにわけて例示されているが、
日本では薬事手続は2つのみ。また、年次報告の仕組みもない。
⚫ なお、以下の表は、ICH Q12の分類に従って3極の分類を当てはめたものであり、実際の変更事項の分類が3極で一致するものではない。
ICH Q12の分類
事前承認
届出・中リスク
米国
EU
日本
PAS(Prior Approval Supplement )
Type II Variation
一部変更承認申請
変更前に事前申請
変更前に事前申請
変更前に事前申請
CBE30
Type IB Variation
変更計画を提出し、受領連絡(提出から
14日以内)から30日以内に連絡がなけ
れば変更可
変更計画を提出し、受領連絡(提出から
7日程度)から30日以内に連絡がなけれ
ば変更可
中リスクに対応する
カテゴリがない
Type IAIN Variation
変更計画を提出し、受領連絡後に変更可
変更後、速やかに変更内容を提出。有効
or無効のフィードバックが30日以内にあ
る。
Annual Report
Type IA Variation
変更事項を1年に1回提出
変更後12か月以内に変更内容を提出。他
の変更と併せて、年次報告とすることも
可能。
CBE0
届出・低リスク
軽微変更届出
変更後30日以内に届出
※実際には欧米では届出相当の手続も日本
では軽微変更ではなく一部変更申請が求め
られる場合がある(次ページ以降参照)
Annual Report、Type IA Valiation
のような、年次報告の仕組みがない
報告不要
3
図9 日米欧の変更管理の手続の概要
(3)対応の方向性
①
中等度変更事項の導入
医薬品の製造方法等の変更管理については、欧米と同様に、変更案を提出し、短
期間の確認期間を経て変更を行うことができる新たな変更カテゴリとして「中等度
変更事項」を導入すべきこととした。
制度の詳細やフィージビリティを検討するため、まずは対象を限定して試行的に
導入すべきこととした。
試行における「中等度変更事項」の対象については、変更内容のリスクの程度に
基づき、①初回承認申請又は一変申請の審査においてあらかじめ「中等度変更事項」
として特定された事項、及び②変更が生じた都度の PMDA 相談で中等度変更事項へ
の該当性を確認された事項とした。
また、試行においての「中等度変更事項」に係る薬事手続は、現行の一変申請の
一類型とした上で、その審査を短期間で実施することとした。
19
https://www.mhlw.go.jp/content/11121000/001155992.pdf
20
https://www.mhlw.go.jp/content/11121000/001155993.pdf
30