


























よむ、つかう、まなぶ。
参考資料 2 血漿分画製剤のウイルスに対する安全性確保に関するガイドラインの一部改正について(令和6年3月 29 日付け医薬発 0329 第 16 号厚生労働省医薬局長通知) (24 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_40647.html |
| 出典情報 | 薬事審議会 血液事業部会運営委員会(令和6年度第1回 6/19)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
は同じ特性を持っているなどの理由で 2 種類のモデルウイルスを選択することが可能な場合には、原則と 照。
してウイルス除去及び不活化処理に対してより抵抗性の強いウイルスを選択すること。
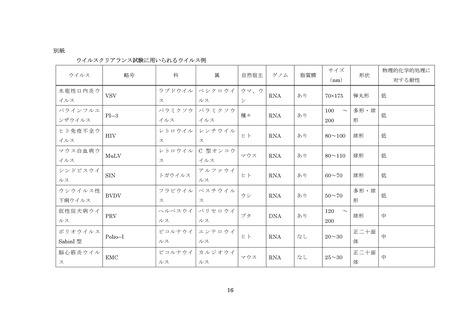
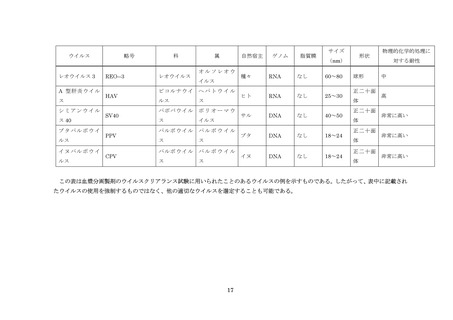
血漿分画製剤のウイルスクリアランス試験に用いられるウイルスの例については別紙を参照すること。
4
ウイルスクリア
ランス試験
4.3 ウイルスクリアランス試験の設計
4.3 ウイルス・プロセスバリデーション試験の設計
ウイルスクリアランス試験は、対象となる特定の製造工程段階で意図的にウイルスを添加し、当該製造
ウイルス・プロセスバリデーション試験は、対象となる特定の製造工程段階で意図的にウイルスを添加
工程のウイルスクリアランス能を定量的に評価するものである。したがって、当該製剤の全ての製造工程 し、当該製造工程のウイルス除去及び不活化の能力を定量的に評価するものである。したがって、当該製
を検証する必要はなく、ウイルスの除去及び不活化に寄与する製造工程だけについて実施すること。
剤の全ての製造工程を検証する必要はなく、ウイルスの除去及び不活化に寄与する製造工程だけについて
ウイルスクリアランス能の評価においては、製造者がその製造工程を適切に反映した実験室規模で実施 実施する。
した結果に基づいて評価することを原則とする。いかなるウイルスも製造施設に故意に持ち込むことはで
バリデーションデータは、製造者がその製造工程を縮小した規模で実施した結果に基づいて作成したも
きないため、ウイルスクリアランス試験は、製造設備とは別のウイルス試験設備で行わなければならない。 のを原則として使用する。いかなるウイルスも製造施設に故意に持ち込むことはできないため、バリデー
このため、ウイルスクリアランス試験は、ウイルス学的研究を行う設備のある隔離されたウイルス試験設 ション試験は、製造設備とは別のウイルス試験設備で行わなければならない。このため、バリデーション
備において、ウイルス学の専門家と生産技術者が共同で行う必要がある。この製造規模を縮小して行うウ は、ウイルス学的研究を行う設備のある隔離された別の施設においてウイルス学の専門家と生産技術者が
イルスクリアランス試験は、実生産規模での製造工程との同等性が検証されていることが前提でなければ 共同で行う必要がある。この製造規模を縮小して行うバリデーション試験は、実生産規模での製造工程と
ならない。クロマトグラフィー工程については、カラムベッド高、線流速、ベッド容量に対する流速の比 の同等性が検証されていることが前提でなければならない。クロマトグラフ装置については、カラムベッ
率(すなわち接触時間)
、緩衝液、カラム充填剤の種類、pH、温度、タンパク質濃度、塩濃度、製品濃度 ト高、線流速、ベット容量に対する流速の比率(すなわち接触時間)、緩衝液、カラム充填剤の種類、pH、
に関しても、全て実生産スケールの製造に対応している必要がある。また、溶出のプロフィールも同様の 温度、たん白濃度、塩濃度、製品濃度に関しても、全て実生産スケールの製造に対応している必要がある。
ものが得られるように設計するべきである。同様な考え方をその他の工程についても適用することが必要 また、溶出のプロフィールも同様のものが得られるように設計するべきである。同様な考え方をその他の
である。しかし、やむを得ない事情により実際の製造工程を反映させることができない場合には、それが 工程についても適用することが必要である。しかし、やむを得ない事情により実際の製造工程を反映させ
結果にどの様な影響を及ぼすかを考察しておくべきである。
ることができない場合には、それが結果にどの様な影響を及ぼすかを考察しておくべきである。
ウイルスクリアランス試験の計画を立案する際、検討することが望ましい留意点を以下に示す。
ウイルス・プロセスバリデーション試験の計画を立案する際、検討することが望ましい留意点を以下に
示す。
(1)製造工程の設計にあたっては、ウイルスを除去又は不活化できる、機序の異なる 2 つ以上の工程の (1) 製造工程の設計には 2 つ以上の異なるウイルス不活化及び除去工程について検討することが望まし
採用について検討するように努めること。
い。
(2)ウイルスを除去又は不活化することが予想される工程について、その能力を個々に評価し、それぞれ (2) ウイルスを不活化及び除去することが予想される工程について、その能力を個々に評価し、それぞ
が除去工程なのか、不活化工程なのか、あるいは除去及び不活化のいずれにも関与しているものかを
れが不活化工程なのか、除去工程なのか、あるいは不活化・除去いずれにも関与しているものかを明ら
明らかにできるような試験を計画すること。
かにできるような試験を計画すべきである。
(3)ウイルスクリアランス能に影響を及ぼす製造工程上の変動因子について検討すること。
(3) ウイルス除去及び不活化効果に影響を及ぼす製造工程上の変動因子について検討する。
(4)ウイルスに対する抗体が出発原料に存在する場合には、ウイルス除去工程及び不活化工程における (4) ウイルスに対する抗体が出発原料に存在する場合には、ウイルス除去及び不活化工程におけるウイ
ウイルスの挙動に影響を及ぼす可能性があるので、ウイルスクリアランス試験ではこのことを考慮し
ルスの挙動に影響を及ぼす可能性があるので、バリデーション試験ではこのことを考慮して実施する。
て実施する。また、原血漿の混合により抗体が特定のウイルスの不活化に寄与することを評価する場
合には、抗体の中和活性を適切に評価できるアッセイ法を用いる必要がある。
(5)試料中に添加するウイルス量は、その製造工程のウイルスクリアランス能を充分に評価できる量と (5) 試料中に添加するウイルス量は、その製造工程のウイルス除去及び不活化能力を充分に評価できる
する。ただし、一般的にウイルスの添加量は、ウイルス溶液量として出発原料の 10%以下とすること。 量とする。ただし、一般的にウイルスの添加量はウイルスの溶液量として出発原料の 10%以下とするこ
とが望ましい。
(6)試料中のウイルスは、可能な限り超遠心分離、透析、保存などの操作を行わずに定量することが望ま (6) 試料中のウイルスは、可能な限り超遠心分離、透析、保存などの操作を行わずに定量することが望ま
しい。しかし、試験に対する阻害物質や使用する細胞に対する毒性物質を除去するため、又は全ての
しい。しかし、阻害物質や毒性物質を除去するため、又は全ての試料を同時に定量するため、定量前に
試料を同時に定量するため、定量前に何らかの処理をすることが避けられない場合には、適切なコン
何らかの処理をすることが避けられない場合には、適切なコントロールを用いて、その処理の試験結果
トロールを用いて、その処理の試験結果に対する影響を確認するとともに、試料による毒性発現など
に対する影響を確認するとともに、試料による毒性発現などの検出系に対する影響も考慮する。
の検出系に対する影響も考慮すること。
(7)ウイルスの選択にあたっては、ウイルスクリアランス試験従事者に健康被害をもたらす可能性のあ (7) ウイルスの選択にあたっては、クリアランス試験従事者に健康被害をもたらす可能性のあることに
ることに配慮すること。
配慮するべきである。
また、ウイルス・プロセスバリデーション試験は「医薬品及び医薬部外品の製造管理及び品質管理規
則(H11 年 3 月 12 日厚生省令第 16 号)」の手続きに基づいて、実施しなければならない。
4.4 ウイルスクリアランス能の評価
4.4.1 ウイルスクリアランス指数の評価
製造工程におけるウイルスクリアランス指数は、各製造段階での試験で得られたウイルスクリアランス
指数の総和で評価する。製造販売業者は、得られたウイルスクリアランス指数が適切であるかどうかにつ
いて、原血漿及び製造過程に含まれる可能性のある全てのウイルスを念頭において評価し、その妥当性を
示すべきである。
4.4 ウイルス・プロセスバリデーションの評価
4.4.1 ウイルス低減率(ウイルスクリアランス指数)の評価
製造工程の各製造段階でのウイルスクリアランス指数は、ウイルス・プロセスバリデーション試験の結
果により得られたウイルス減少値の総和で評価する。製造業者は、得られたウイルスクリアランス指数が
受け入れ可能かどうかについて、原料血漿及び製造過程に含まれる可能性のある全てのウイルスを念頭に
おいて評価し、その妥当性を示すべきである。
5
24
してウイルス除去及び不活化処理に対してより抵抗性の強いウイルスを選択すること。
血漿分画製剤のウイルスクリアランス試験に用いられるウイルスの例については別紙を参照すること。
4
ウイルスクリア
ランス試験
4.3 ウイルスクリアランス試験の設計
4.3 ウイルス・プロセスバリデーション試験の設計
ウイルスクリアランス試験は、対象となる特定の製造工程段階で意図的にウイルスを添加し、当該製造
ウイルス・プロセスバリデーション試験は、対象となる特定の製造工程段階で意図的にウイルスを添加
工程のウイルスクリアランス能を定量的に評価するものである。したがって、当該製剤の全ての製造工程 し、当該製造工程のウイルス除去及び不活化の能力を定量的に評価するものである。したがって、当該製
を検証する必要はなく、ウイルスの除去及び不活化に寄与する製造工程だけについて実施すること。
剤の全ての製造工程を検証する必要はなく、ウイルスの除去及び不活化に寄与する製造工程だけについて
ウイルスクリアランス能の評価においては、製造者がその製造工程を適切に反映した実験室規模で実施 実施する。
した結果に基づいて評価することを原則とする。いかなるウイルスも製造施設に故意に持ち込むことはで
バリデーションデータは、製造者がその製造工程を縮小した規模で実施した結果に基づいて作成したも
きないため、ウイルスクリアランス試験は、製造設備とは別のウイルス試験設備で行わなければならない。 のを原則として使用する。いかなるウイルスも製造施設に故意に持ち込むことはできないため、バリデー
このため、ウイルスクリアランス試験は、ウイルス学的研究を行う設備のある隔離されたウイルス試験設 ション試験は、製造設備とは別のウイルス試験設備で行わなければならない。このため、バリデーション
備において、ウイルス学の専門家と生産技術者が共同で行う必要がある。この製造規模を縮小して行うウ は、ウイルス学的研究を行う設備のある隔離された別の施設においてウイルス学の専門家と生産技術者が
イルスクリアランス試験は、実生産規模での製造工程との同等性が検証されていることが前提でなければ 共同で行う必要がある。この製造規模を縮小して行うバリデーション試験は、実生産規模での製造工程と
ならない。クロマトグラフィー工程については、カラムベッド高、線流速、ベッド容量に対する流速の比 の同等性が検証されていることが前提でなければならない。クロマトグラフ装置については、カラムベッ
率(すなわち接触時間)
、緩衝液、カラム充填剤の種類、pH、温度、タンパク質濃度、塩濃度、製品濃度 ト高、線流速、ベット容量に対する流速の比率(すなわち接触時間)、緩衝液、カラム充填剤の種類、pH、
に関しても、全て実生産スケールの製造に対応している必要がある。また、溶出のプロフィールも同様の 温度、たん白濃度、塩濃度、製品濃度に関しても、全て実生産スケールの製造に対応している必要がある。
ものが得られるように設計するべきである。同様な考え方をその他の工程についても適用することが必要 また、溶出のプロフィールも同様のものが得られるように設計するべきである。同様な考え方をその他の
である。しかし、やむを得ない事情により実際の製造工程を反映させることができない場合には、それが 工程についても適用することが必要である。しかし、やむを得ない事情により実際の製造工程を反映させ
結果にどの様な影響を及ぼすかを考察しておくべきである。
ることができない場合には、それが結果にどの様な影響を及ぼすかを考察しておくべきである。
ウイルスクリアランス試験の計画を立案する際、検討することが望ましい留意点を以下に示す。
ウイルス・プロセスバリデーション試験の計画を立案する際、検討することが望ましい留意点を以下に
示す。
(1)製造工程の設計にあたっては、ウイルスを除去又は不活化できる、機序の異なる 2 つ以上の工程の (1) 製造工程の設計には 2 つ以上の異なるウイルス不活化及び除去工程について検討することが望まし
採用について検討するように努めること。
い。
(2)ウイルスを除去又は不活化することが予想される工程について、その能力を個々に評価し、それぞれ (2) ウイルスを不活化及び除去することが予想される工程について、その能力を個々に評価し、それぞ
が除去工程なのか、不活化工程なのか、あるいは除去及び不活化のいずれにも関与しているものかを
れが不活化工程なのか、除去工程なのか、あるいは不活化・除去いずれにも関与しているものかを明ら
明らかにできるような試験を計画すること。
かにできるような試験を計画すべきである。
(3)ウイルスクリアランス能に影響を及ぼす製造工程上の変動因子について検討すること。
(3) ウイルス除去及び不活化効果に影響を及ぼす製造工程上の変動因子について検討する。
(4)ウイルスに対する抗体が出発原料に存在する場合には、ウイルス除去工程及び不活化工程における (4) ウイルスに対する抗体が出発原料に存在する場合には、ウイルス除去及び不活化工程におけるウイ
ウイルスの挙動に影響を及ぼす可能性があるので、ウイルスクリアランス試験ではこのことを考慮し
ルスの挙動に影響を及ぼす可能性があるので、バリデーション試験ではこのことを考慮して実施する。
て実施する。また、原血漿の混合により抗体が特定のウイルスの不活化に寄与することを評価する場
合には、抗体の中和活性を適切に評価できるアッセイ法を用いる必要がある。
(5)試料中に添加するウイルス量は、その製造工程のウイルスクリアランス能を充分に評価できる量と (5) 試料中に添加するウイルス量は、その製造工程のウイルス除去及び不活化能力を充分に評価できる
する。ただし、一般的にウイルスの添加量は、ウイルス溶液量として出発原料の 10%以下とすること。 量とする。ただし、一般的にウイルスの添加量はウイルスの溶液量として出発原料の 10%以下とするこ
とが望ましい。
(6)試料中のウイルスは、可能な限り超遠心分離、透析、保存などの操作を行わずに定量することが望ま (6) 試料中のウイルスは、可能な限り超遠心分離、透析、保存などの操作を行わずに定量することが望ま
しい。しかし、試験に対する阻害物質や使用する細胞に対する毒性物質を除去するため、又は全ての
しい。しかし、阻害物質や毒性物質を除去するため、又は全ての試料を同時に定量するため、定量前に
試料を同時に定量するため、定量前に何らかの処理をすることが避けられない場合には、適切なコン
何らかの処理をすることが避けられない場合には、適切なコントロールを用いて、その処理の試験結果
トロールを用いて、その処理の試験結果に対する影響を確認するとともに、試料による毒性発現など
に対する影響を確認するとともに、試料による毒性発現などの検出系に対する影響も考慮する。
の検出系に対する影響も考慮すること。
(7)ウイルスの選択にあたっては、ウイルスクリアランス試験従事者に健康被害をもたらす可能性のあ (7) ウイルスの選択にあたっては、クリアランス試験従事者に健康被害をもたらす可能性のあることに
ることに配慮すること。
配慮するべきである。
また、ウイルス・プロセスバリデーション試験は「医薬品及び医薬部外品の製造管理及び品質管理規
則(H11 年 3 月 12 日厚生省令第 16 号)」の手続きに基づいて、実施しなければならない。
4.4 ウイルスクリアランス能の評価
4.4.1 ウイルスクリアランス指数の評価
製造工程におけるウイルスクリアランス指数は、各製造段階での試験で得られたウイルスクリアランス
指数の総和で評価する。製造販売業者は、得られたウイルスクリアランス指数が適切であるかどうかにつ
いて、原血漿及び製造過程に含まれる可能性のある全てのウイルスを念頭において評価し、その妥当性を
示すべきである。
4.4 ウイルス・プロセスバリデーションの評価
4.4.1 ウイルス低減率(ウイルスクリアランス指数)の評価
製造工程の各製造段階でのウイルスクリアランス指数は、ウイルス・プロセスバリデーション試験の結
果により得られたウイルス減少値の総和で評価する。製造業者は、得られたウイルスクリアランス指数が
受け入れ可能かどうかについて、原料血漿及び製造過程に含まれる可能性のある全てのウイルスを念頭に
おいて評価し、その妥当性を示すべきである。
5
24