













































よむ、つかう、まなぶ。
(参考資料3)業界からの要望事項 (17 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_39786.html |
| 出典情報 | 厚生科学審議会 医薬品医療機器制度部会(令和6年度第1回 4/18)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
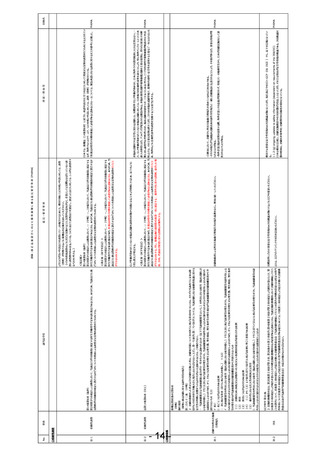
安-2
安-1
No.
使用の成績
安-4
安-5 未知非重篤定期報告
使用の成績
法第14条の4第6項および第14条の5第2項(安全性定期報告)
法施行規則第228条の20第1項第3号(未知非重篤定期報告)
法第14条の4第6項(同上)
法第14条の4第6項
第四項の規定による確認においては、第一項各号に掲げる医薬品に係る申請内容及び前項前段に規定する資料に基づき、当該医薬品の品質、
有効性及び安全性に関する調査を行うものとする。この場合において、第一項各号に掲げる医薬品が前項後段に規定する厚生労働省令で定める
医薬品であるときは、あらかじめ、当該医薬品に係る資料が同項後段の規定に適合するかどうかについての書面による調査又は実地の調査を行うも
のとする。
法第14条の4第4項
承認条件もしくはGVP上の位置づけであるRMPの観点では追加の安全性監視計画(PVP)の一部としてGPSP規制下でのPMSが
厚生労働大臣の再審査は、再審査を行う際に得られている知見に基づき、第一項各号に掲げる医薬品が第十四条第二項第三号イからハまでのい
存在し、入れ子状態となっている。再審査時には通常のPVP情報も含め資料として取りまとめ提出するが、これはGPSPがGVP規制下
GPSPを第十二条(再審査等の資料の基準)のみとし、PMSに関する規定をGVPの安全監視の一部として統
ずれにも該当しないことを確認することにより行う。
の情報についても管理する「逆入れ子状態」となっている。このような状況をクリアするためにGPSPを再審査に限定した枠組みとし、
PhRMA
合する。
PVPに関する部分をGVPに統合する。これによりICH E2Eの観点からも整合すると思われる。
厚生労働省告示第171号康生労働症例第87号(GPSP省令)第十二条(製造販売後調査等に係る再審査等の資料の基準)
使用の成績
再審査期間中の未知非重篤定期報告書を不要とし、未知非重篤事象については安全性定期報告に含めて
報告する。
医薬品の安全性は未知既知、重篤性に関わらずライフサイクルを通して包括的に評価すべきであり、未知非重篤事象を切り出して部
分的に評価することは安全監視上適切ではないと考える。また、再審査期間終了後は、未知非重篤定期報告書のみを継続して提
PhRMA
出が必要となるが、多くの場合安全性プロファイルは確定しているため、新たな知見を得られることもなく、安全性監視に寄与していない
と考えるため。
製造販売後臨床試験を実施する場合を除き、使用の成績に関する調査によって、承認時と同程度のエビデンスレベルの有効性を評
価できることはない。この条文により、RMPの1.2項有効性に関する検討事項がなしとなっているにもかかわらず、追加の医薬品安全性
監視活動の中で有効性評価項目の情報収集が規定するようPMDAから指示される現状がある。臨床試験で設定した有効性の評価
項目が実診療で実施されるケースは非常に限られており、観察研究でのデータ収集は現実的ではない。にもかかわらず、データを収集
し、限られた情報の中で有効性が市販後でも認められていたかのような評価を実施している現状がある。
「第四項の規定による確認においては、第一項各号に掲げる医薬品に係る申請内容及び前項前段に規定する 薬生薬審発0314第4号薬生安発0314第4号「医薬品の製造販売後調査等の実施計画の策定に関する検討の進め方について」
資料に基づき、当該医薬品の品質、有効性及び安全性に関する調査を行うものとする。」とあるが、通知の記載 通知の中で,「一般的に、製造販売承認(以下「承認」という。)に足る有効性に係る情報は、 承認時に評価された治験により収 PhRMA
とも一貫性をそろえて、必ず市販後の有効性評価が必要と読み取れないように記載修正する。
集されており、 承認時に有効性に係る一定の検証がなされている。したがって、承認審査の過程 及び 製造販売後に生じた 評価 す
べき具体的な検討事項が存在しない場合は、製造販売後の有効性評価については、製造販売後調査等によらない 、文献の分析
等 による 方法で 検討すること差し支えない 。一方 、承認審査の過程 及び 製造販売後に、 有効性に関する具体的 な検討事項
が生じた場合は、 当該事項を科学的に確認することが可能となる製造販売後調査等を実施する必要がある。」
この通知の記載通りの運用がされておらず,その原因が薬機法の指摘箇所にあると考えるため,必ず市販後の有効性評価必要と読
みとれてしまう今の方の記載の修正を検討して頂きたい。
今般、「創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会」で、使用成績調査に関する企業や医療機
関への過度な負担が指摘されている。これは本条文に起因する局⾧通知(薬食発第1220008号)に定めた内容の実施の有無を
「この場合において、第一項各号に掲げる医薬品が前項後段に規定する厚生労働省令で定める医薬品であると 確認する作業のために、企業、医療機関、PMDAの業務を規定したためである。また、臨床試験においてはリスクベースドアプローチによ
きは、あらかじめ、当該医薬品に係る資料が同項後段の規定に適合するかどうかについての書面による調査又は る試験目的に適合した品質管理が議論されている。
実地の調査を行うものとする。」の条文の削除
一方、使用成績調査は日本国内でのみ実施されている背景もあり、欧米と調和したICH E6のような指針がないため、欧米と調和の
とれた品質管理の考え方が導入されていない現状がある。使用成績調査では原資料との整合性確認ができない制限の中、局⾧通 PhRMA
改めて、医薬品のリスク管理を行うという本来の目的を考え情報の信頼性保証をすべきであり、製造販売後に実 知(薬食発第1220008号)に「調査票に必須事項が記載されていることの確認」という記載があり、例えば、計画した通り必須事
施される観察研究に対して現行のGPSP省令で規定している信頼性の確認を再審査で求める必要はないと考え 項が記載されているか、記載がない場合は本当に情報がないのか医療機関に問い合わせるという不毛な業務も発生している。
る。
本条文は、3000例の使用成績調査をどの医薬品でも実施させてきた過去の想定を基に作られたものであり、リスク管理計画に基づく
安全性監視に切り替わった現代においては不要と考える。再審査に提出する使用の成績に関連する調査はリスク管理が主目的であり
(RMPの実施結果の確認)、使用の成績に関する調査が省令通り実施されていたかどうかは企業の自主担保でよいと考える。
「第一項各号に掲げる医薬品につき第十四条の承認を受けた者は、厚生労働省令で定めるところにより、当該
医薬品のリスク管理計画を実施し、その結果を厚生労働大臣に報告しなければならない。」と修正し、「使用の成
PhRMA
績に関する調査その他厚生労働省令で定める調査」が必須と読める条文を改定する。
参考:薬生薬審発0314第4号、薬生安発0314 第4号「医薬品の製造販売後調査等の実施計画の策定に関する検討の進め方
について」では、追加のPVPが不要な承認も想定される。
参考:市販後の日本の安全性監視にかかる業務の予測可能性が低いことがドラッグロス・ラグ問題を引き起こす可能性がある。また、
日本の再審査制度を参考にして同制度を導入した韓国において、再審査制度を廃止し、RMP制度への切り替えが発表された
(https://www.mfds.go.kr/brd/m_99/view.do?seq=44429)。
参考:法施行規則第五十九条に以下の規定があるが、実際そのように運用されている事例は限定的である。また合理的な理由につ
いての基準が明確ではなく苦慮している。
「法第十四条の四第五項の規定により第五十六条の申請書に添付しなければならない資料は、申請に係る医薬品の使用成績に関
する資料、第六十三条第二項の規定による報告に際して提出した資料の概要その他当該医薬品の効能又は効果及び安全性に関
しその製造販売の承認後に得られた研究報告に関する資料とする。ただし、使用成績に関する資料については、添付を必要としない
合理的理由がある場合は、この限りでない。」
団体名
法第14条の4第7項
第一項各号に掲げる医薬品につき第十四条の承認を受けた者は、厚生労働省令で定めるところにより、当該医薬品の使用の成績に関する調査そ
の他厚生労働省令で定める調査を行い、その結果を厚生労働大臣に報告しなければならない。
法第23条の26第3項、第23条の29第6項(省略)
本邦においては、平成24年4月に医薬品安全性監視計画に加えて、医薬品のリスクの低減を図るためのリスク最小化計画を含めた
医薬品リスク管理計画(RMP: Risk Management Plan)を策定するための指針がとりまとめられ、承認後の医薬品のリスク管理
が行われている。
現在の実態に合わせて、薬機法の「使用の成績に関する調査」を実施させる規制から、リスク管理として必要な活動を実施し、医薬品
のベネフィットリスクバランスの評価を行い報告させる規制に改正が必要である。RMPやそのも元なるICH E2Eに従った医薬品安全性
監視の計画は欧米で標準的に実施されている。
日本においては、この条文により欧米で実施している医薬品安全性監視に加え、日本で「使用の成績に関する調査」の実施が必要と
なっており、海外企業の日本での医薬品開発の参入障壁になっている可能性がある。近年、画期的な新薬は国際共同で同時開発さ
れ、世界同時承認を進める企業が多くなっている。これは、承認後に世界で同時に安全性情報を収集し、リスク管理をしていく体制構
築が必要になることを意味する。このような背景のもと、本邦において独自の「使用の成績」に関する調査を実施させる規制ではなく、国
際調和した市販後のリスク管理をさせるよう規制改革するべきであると考える。
背景・理由等
使用の成績
意見・要望事項
該当法令等
事項
安-3
- 16 -
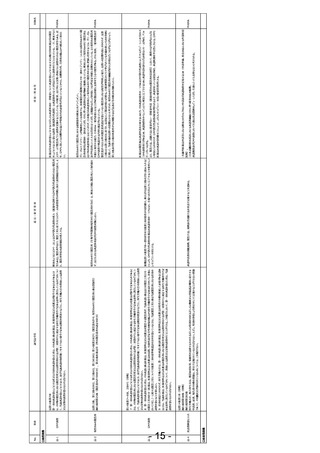
安-1
No.
使用の成績
安-4
安-5 未知非重篤定期報告
使用の成績
法第14条の4第6項および第14条の5第2項(安全性定期報告)
法施行規則第228条の20第1項第3号(未知非重篤定期報告)
法第14条の4第6項(同上)
法第14条の4第6項
第四項の規定による確認においては、第一項各号に掲げる医薬品に係る申請内容及び前項前段に規定する資料に基づき、当該医薬品の品質、
有効性及び安全性に関する調査を行うものとする。この場合において、第一項各号に掲げる医薬品が前項後段に規定する厚生労働省令で定める
医薬品であるときは、あらかじめ、当該医薬品に係る資料が同項後段の規定に適合するかどうかについての書面による調査又は実地の調査を行うも
のとする。
法第14条の4第4項
承認条件もしくはGVP上の位置づけであるRMPの観点では追加の安全性監視計画(PVP)の一部としてGPSP規制下でのPMSが
厚生労働大臣の再審査は、再審査を行う際に得られている知見に基づき、第一項各号に掲げる医薬品が第十四条第二項第三号イからハまでのい
存在し、入れ子状態となっている。再審査時には通常のPVP情報も含め資料として取りまとめ提出するが、これはGPSPがGVP規制下
GPSPを第十二条(再審査等の資料の基準)のみとし、PMSに関する規定をGVPの安全監視の一部として統
ずれにも該当しないことを確認することにより行う。
の情報についても管理する「逆入れ子状態」となっている。このような状況をクリアするためにGPSPを再審査に限定した枠組みとし、
PhRMA
合する。
PVPに関する部分をGVPに統合する。これによりICH E2Eの観点からも整合すると思われる。
厚生労働省告示第171号康生労働症例第87号(GPSP省令)第十二条(製造販売後調査等に係る再審査等の資料の基準)
使用の成績
再審査期間中の未知非重篤定期報告書を不要とし、未知非重篤事象については安全性定期報告に含めて
報告する。
医薬品の安全性は未知既知、重篤性に関わらずライフサイクルを通して包括的に評価すべきであり、未知非重篤事象を切り出して部
分的に評価することは安全監視上適切ではないと考える。また、再審査期間終了後は、未知非重篤定期報告書のみを継続して提
PhRMA
出が必要となるが、多くの場合安全性プロファイルは確定しているため、新たな知見を得られることもなく、安全性監視に寄与していない
と考えるため。
製造販売後臨床試験を実施する場合を除き、使用の成績に関する調査によって、承認時と同程度のエビデンスレベルの有効性を評
価できることはない。この条文により、RMPの1.2項有効性に関する検討事項がなしとなっているにもかかわらず、追加の医薬品安全性
監視活動の中で有効性評価項目の情報収集が規定するようPMDAから指示される現状がある。臨床試験で設定した有効性の評価
項目が実診療で実施されるケースは非常に限られており、観察研究でのデータ収集は現実的ではない。にもかかわらず、データを収集
し、限られた情報の中で有効性が市販後でも認められていたかのような評価を実施している現状がある。
「第四項の規定による確認においては、第一項各号に掲げる医薬品に係る申請内容及び前項前段に規定する 薬生薬審発0314第4号薬生安発0314第4号「医薬品の製造販売後調査等の実施計画の策定に関する検討の進め方について」
資料に基づき、当該医薬品の品質、有効性及び安全性に関する調査を行うものとする。」とあるが、通知の記載 通知の中で,「一般的に、製造販売承認(以下「承認」という。)に足る有効性に係る情報は、 承認時に評価された治験により収 PhRMA
とも一貫性をそろえて、必ず市販後の有効性評価が必要と読み取れないように記載修正する。
集されており、 承認時に有効性に係る一定の検証がなされている。したがって、承認審査の過程 及び 製造販売後に生じた 評価 す
べき具体的な検討事項が存在しない場合は、製造販売後の有効性評価については、製造販売後調査等によらない 、文献の分析
等 による 方法で 検討すること差し支えない 。一方 、承認審査の過程 及び 製造販売後に、 有効性に関する具体的 な検討事項
が生じた場合は、 当該事項を科学的に確認することが可能となる製造販売後調査等を実施する必要がある。」
この通知の記載通りの運用がされておらず,その原因が薬機法の指摘箇所にあると考えるため,必ず市販後の有効性評価必要と読
みとれてしまう今の方の記載の修正を検討して頂きたい。
今般、「創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会」で、使用成績調査に関する企業や医療機
関への過度な負担が指摘されている。これは本条文に起因する局⾧通知(薬食発第1220008号)に定めた内容の実施の有無を
「この場合において、第一項各号に掲げる医薬品が前項後段に規定する厚生労働省令で定める医薬品であると 確認する作業のために、企業、医療機関、PMDAの業務を規定したためである。また、臨床試験においてはリスクベースドアプローチによ
きは、あらかじめ、当該医薬品に係る資料が同項後段の規定に適合するかどうかについての書面による調査又は る試験目的に適合した品質管理が議論されている。
実地の調査を行うものとする。」の条文の削除
一方、使用成績調査は日本国内でのみ実施されている背景もあり、欧米と調和したICH E6のような指針がないため、欧米と調和の
とれた品質管理の考え方が導入されていない現状がある。使用成績調査では原資料との整合性確認ができない制限の中、局⾧通 PhRMA
改めて、医薬品のリスク管理を行うという本来の目的を考え情報の信頼性保証をすべきであり、製造販売後に実 知(薬食発第1220008号)に「調査票に必須事項が記載されていることの確認」という記載があり、例えば、計画した通り必須事
施される観察研究に対して現行のGPSP省令で規定している信頼性の確認を再審査で求める必要はないと考え 項が記載されているか、記載がない場合は本当に情報がないのか医療機関に問い合わせるという不毛な業務も発生している。
る。
本条文は、3000例の使用成績調査をどの医薬品でも実施させてきた過去の想定を基に作られたものであり、リスク管理計画に基づく
安全性監視に切り替わった現代においては不要と考える。再審査に提出する使用の成績に関連する調査はリスク管理が主目的であり
(RMPの実施結果の確認)、使用の成績に関する調査が省令通り実施されていたかどうかは企業の自主担保でよいと考える。
「第一項各号に掲げる医薬品につき第十四条の承認を受けた者は、厚生労働省令で定めるところにより、当該
医薬品のリスク管理計画を実施し、その結果を厚生労働大臣に報告しなければならない。」と修正し、「使用の成
PhRMA
績に関する調査その他厚生労働省令で定める調査」が必須と読める条文を改定する。
参考:薬生薬審発0314第4号、薬生安発0314 第4号「医薬品の製造販売後調査等の実施計画の策定に関する検討の進め方
について」では、追加のPVPが不要な承認も想定される。
参考:市販後の日本の安全性監視にかかる業務の予測可能性が低いことがドラッグロス・ラグ問題を引き起こす可能性がある。また、
日本の再審査制度を参考にして同制度を導入した韓国において、再審査制度を廃止し、RMP制度への切り替えが発表された
(https://www.mfds.go.kr/brd/m_99/view.do?seq=44429)。
参考:法施行規則第五十九条に以下の規定があるが、実際そのように運用されている事例は限定的である。また合理的な理由につ
いての基準が明確ではなく苦慮している。
「法第十四条の四第五項の規定により第五十六条の申請書に添付しなければならない資料は、申請に係る医薬品の使用成績に関
する資料、第六十三条第二項の規定による報告に際して提出した資料の概要その他当該医薬品の効能又は効果及び安全性に関
しその製造販売の承認後に得られた研究報告に関する資料とする。ただし、使用成績に関する資料については、添付を必要としない
合理的理由がある場合は、この限りでない。」
団体名
法第14条の4第7項
第一項各号に掲げる医薬品につき第十四条の承認を受けた者は、厚生労働省令で定めるところにより、当該医薬品の使用の成績に関する調査そ
の他厚生労働省令で定める調査を行い、その結果を厚生労働大臣に報告しなければならない。
法第23条の26第3項、第23条の29第6項(省略)
本邦においては、平成24年4月に医薬品安全性監視計画に加えて、医薬品のリスクの低減を図るためのリスク最小化計画を含めた
医薬品リスク管理計画(RMP: Risk Management Plan)を策定するための指針がとりまとめられ、承認後の医薬品のリスク管理
が行われている。
現在の実態に合わせて、薬機法の「使用の成績に関する調査」を実施させる規制から、リスク管理として必要な活動を実施し、医薬品
のベネフィットリスクバランスの評価を行い報告させる規制に改正が必要である。RMPやそのも元なるICH E2Eに従った医薬品安全性
監視の計画は欧米で標準的に実施されている。
日本においては、この条文により欧米で実施している医薬品安全性監視に加え、日本で「使用の成績に関する調査」の実施が必要と
なっており、海外企業の日本での医薬品開発の参入障壁になっている可能性がある。近年、画期的な新薬は国際共同で同時開発さ
れ、世界同時承認を進める企業が多くなっている。これは、承認後に世界で同時に安全性情報を収集し、リスク管理をしていく体制構
築が必要になることを意味する。このような背景のもと、本邦において独自の「使用の成績」に関する調査を実施させる規制ではなく、国
際調和した市販後のリスク管理をさせるよう規制改革するべきであると考える。
背景・理由等
使用の成績
意見・要望事項
該当法令等
事項
安-3
- 16 -