























よむ、つかう、まなぶ。
【資料2】テーマ②(新技術による医薬品等にも対応したリスクに基づく市販後安全性対策の強化、法違反事例を踏まえた更なる法令遵守や品質確保の取組の実施)について.pdf (4 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_41209.html |
| 出典情報 | 厚生科学審議会 医薬品医療機器制度部会(令和6年度第4回 7/5)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
参考
RMP制度と運用実態について
○
RMPは承認時だけでなく、製造販売後に新たな安全性上の懸念が判明した場合も作成することとなっている。しかしな
がら、実態は、新薬承認時に承認条件として作成され、再審査時にRMPに基づく活動の実施内容を確認し、承認条件を解
除するという運用がほとんど。RMPを作成していない医薬品で、製造販売後にRMP策定を義務付けた事例は限られる。
○
また、承認条件が解除されるまでのRMPの変更について、状況のアップデートが多く、リスク最小化策の効果評価や潜
在リスクの見直し等が行われていないことが指摘されている。
(参考)欧州では2017年3月にガイドラインが見直され、「安全性検討事項は、一般に、リスクの中でも追加の安全性監視や追加のリスク最小化策が
必要となるもの」との考えが示された結果、製造販売後の情報に基づき安全性検討事項が柔軟に変更されるようになったとの報告※がある。
※Impact of Changing Regulations and the Dynamic Nature of European Risk Management Plans for
Human Medicines on the Lifecycle of Safety Concerns, Pharmaceutical Medicine (2022) 36:33–46
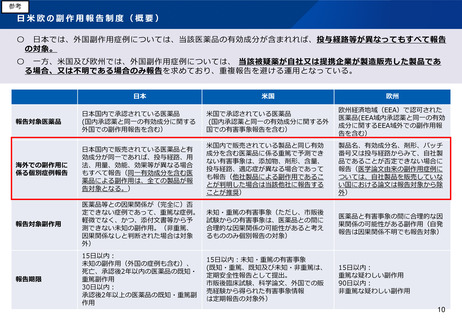
日米欧のRMP制度(概要)
製造販売後にRMPを承認条件として設定した例
日本
欧州
米国
制度名
医薬品リスク管理計
画(RMP)
Risk Management
Plan(RMP)
Risk Evaluation and
Mitigation
Strategies(REMS)
対象
・新医薬品
・バイオ後続品
・後発医薬品
・新医薬品
・バイオ後続品
・後発医薬品など
新医薬品および既承
認医薬品のうち、
FDAが必要と判断し
たもの
※企業は承認申請時 ※企業は承認申請時
に案を提出。製販後 に案を提出。製販後
に新たな安全性上の に新たな安全性上の
懸念が判明した場合 懸念が判明した場合
も作成。ただし、承 も作成。策定された
認条件により策定・ RMP全体を取り消す
解除。
仕組みはない。
構成
内容の
変更
・安全性検討事項
・安全性監視計画
・リスク最小化計画
等
・安全性検討事項
・安全性監視計画
・リスク最小化計画
等
ラモトリギン錠の重篤な皮膚
障害による死亡事例の発生を
踏まえ、RMPの策定を義務付
け、追加のリスク最小化計画
として、医療従事者及び患者
向け資材の作成と配布を実施
日本におけるRMP変更状況(RMPが提出された270医薬品の、
2015年8月~2017年3月の状況)
更新内容
件数(%)
・リスク最小化計画
安全上の懸念に関す 安全上の懸念に関す 承認から一定期間後
るエビデンスを踏ま るエビデンスを踏ま に評価した結果によ
え追加、削除、変更。 え追加、削除、変更。 り、変更または削除
市販後の集積情報に基づく安全性検討事項の
変更等(特定リスクへの追加、潜在→特定リ
スクへの変更、不足情報からの削除など)
57(15.2%)
状況のアップデート、記載整備など
318(84.8%)
計
375
AMED研究「RMP制度の効果的な実施と一層の充実のための基礎研究」
平成28年度報告書
4
RMP制度と運用実態について
○
RMPは承認時だけでなく、製造販売後に新たな安全性上の懸念が判明した場合も作成することとなっている。しかしな
がら、実態は、新薬承認時に承認条件として作成され、再審査時にRMPに基づく活動の実施内容を確認し、承認条件を解
除するという運用がほとんど。RMPを作成していない医薬品で、製造販売後にRMP策定を義務付けた事例は限られる。
○
また、承認条件が解除されるまでのRMPの変更について、状況のアップデートが多く、リスク最小化策の効果評価や潜
在リスクの見直し等が行われていないことが指摘されている。
(参考)欧州では2017年3月にガイドラインが見直され、「安全性検討事項は、一般に、リスクの中でも追加の安全性監視や追加のリスク最小化策が
必要となるもの」との考えが示された結果、製造販売後の情報に基づき安全性検討事項が柔軟に変更されるようになったとの報告※がある。
※Impact of Changing Regulations and the Dynamic Nature of European Risk Management Plans for
Human Medicines on the Lifecycle of Safety Concerns, Pharmaceutical Medicine (2022) 36:33–46
日米欧のRMP制度(概要)
製造販売後にRMPを承認条件として設定した例
日本
欧州
米国
制度名
医薬品リスク管理計
画(RMP)
Risk Management
Plan(RMP)
Risk Evaluation and
Mitigation
Strategies(REMS)
対象
・新医薬品
・バイオ後続品
・後発医薬品
・新医薬品
・バイオ後続品
・後発医薬品など
新医薬品および既承
認医薬品のうち、
FDAが必要と判断し
たもの
※企業は承認申請時 ※企業は承認申請時
に案を提出。製販後 に案を提出。製販後
に新たな安全性上の に新たな安全性上の
懸念が判明した場合 懸念が判明した場合
も作成。ただし、承 も作成。策定された
認条件により策定・ RMP全体を取り消す
解除。
仕組みはない。
構成
内容の
変更
・安全性検討事項
・安全性監視計画
・リスク最小化計画
等
・安全性検討事項
・安全性監視計画
・リスク最小化計画
等
ラモトリギン錠の重篤な皮膚
障害による死亡事例の発生を
踏まえ、RMPの策定を義務付
け、追加のリスク最小化計画
として、医療従事者及び患者
向け資材の作成と配布を実施
日本におけるRMP変更状況(RMPが提出された270医薬品の、
2015年8月~2017年3月の状況)
更新内容
件数(%)
・リスク最小化計画
安全上の懸念に関す 安全上の懸念に関す 承認から一定期間後
るエビデンスを踏ま るエビデンスを踏ま に評価した結果によ
え追加、削除、変更。 え追加、削除、変更。 り、変更または削除
市販後の集積情報に基づく安全性検討事項の
変更等(特定リスクへの追加、潜在→特定リ
スクへの変更、不足情報からの削除など)
57(15.2%)
状況のアップデート、記載整備など
318(84.8%)
計
375
AMED研究「RMP制度の効果的な実施と一層の充実のための基礎研究」
平成28年度報告書
4