




























よむ、つかう、まなぶ。
【資料5】創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会 報告書(案) (10 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_38892.html |
| 出典情報 | 創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会(第9回 3/21)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
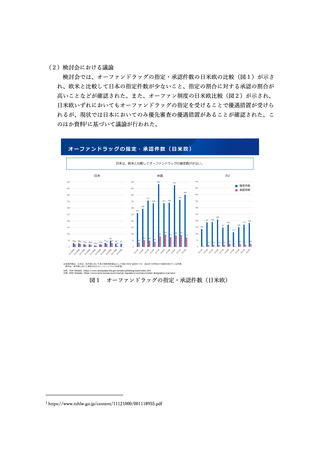
また、小児用医薬品の開発にはコストを要するものの、成人に比べて市場規模は小
さく、コストの回収が困難である。開発コストの低減に資するため、国内で小児の治
験を実施することなく承認申請可能なケースを整理し、明確化すべきと考えられた。
このため、以下のような取組により、小児の治験実施の要否に関する考え方を整理
し、明確化すべきとした。
国際的に用いられているモデリング&シミュレーション(M&S)の活用や、海
外データ、文献情報等により有効性・安全性が説明できる場合を整理し、明確
化する。
新有効成分や新効能医薬品については、少なくとも 10-12 歳以上の小児におい
ては、一定の条件を満たせば、成人の承認申請時に併せて評価可能な場合があ
ること(「成人と合わせて評価可能な小児(10 歳又は 12 歳以上の小児)の臨床
評価の留意点について」
(令和2年6月 30 日厚生労働省医薬・生活衛生局医薬
品審査管理課事務連絡)
)を周知する。
上記に関する相談への対応を含め、PMDA に小児用医薬品に特化した相談枠を新設
すべきとされた。
加えて、例えば、地域において中心的に小児剤形に対応する薬局を設置するなど、
小児剤形を利用しやすくなる仕組みを検討すべきこととした。
(4)検討会後の対応状況
検討会の議論を踏まえ、次の通知を発出した。
・「成人を対象とした医薬品の開発期間中に行う小児用医薬品の開発計画の策定につ
いて」
(令和 6 年 1 月 12 日医薬薬審発 0112 第3号厚生労働省医薬局医薬品審査管
理課長通知)
4.我が国の承認審査における日本人データの必要性の整理(国際共同治験に参加する場
合の日本人第1相試験の必要性)
(1)背景
近年、創薬環境の変化に伴い、大手製薬企業であっても創薬シーズをベンチャー等
の新興バイオ医薬品企業に依存する傾向が強まっている。欧米の新興バイオ医薬品企
業は、経営上の事情等から、開発の早期段階では、日本での開発を欧米と同時に行う
ことは少ないため、大手製薬企業が導入した以降(主に第2相試験の終了後)に日本
での開発を検討・着手するケースが増加している。
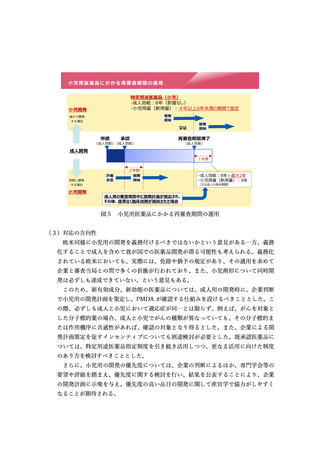
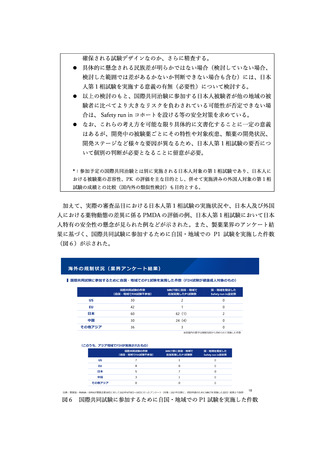
現在、国際共同治験に日本が参加するにあたって、日本人での安全性に関する説明
が十分になされない場合は、事前に日本人での第1相試験を実施する必要があるとさ
れている。一方、日本人第1相試験を実施するためには、一定の時間と費用を要する
ことから、それによって第3相試験の開始が遅延したり、それを回避するために日本
での開発を諦めるケースがあるとの指摘がある。治験における安全性の確保と、新薬
さく、コストの回収が困難である。開発コストの低減に資するため、国内で小児の治
験を実施することなく承認申請可能なケースを整理し、明確化すべきと考えられた。
このため、以下のような取組により、小児の治験実施の要否に関する考え方を整理
し、明確化すべきとした。
国際的に用いられているモデリング&シミュレーション(M&S)の活用や、海
外データ、文献情報等により有効性・安全性が説明できる場合を整理し、明確
化する。
新有効成分や新効能医薬品については、少なくとも 10-12 歳以上の小児におい
ては、一定の条件を満たせば、成人の承認申請時に併せて評価可能な場合があ
ること(「成人と合わせて評価可能な小児(10 歳又は 12 歳以上の小児)の臨床
評価の留意点について」
(令和2年6月 30 日厚生労働省医薬・生活衛生局医薬
品審査管理課事務連絡)
)を周知する。
上記に関する相談への対応を含め、PMDA に小児用医薬品に特化した相談枠を新設
すべきとされた。
加えて、例えば、地域において中心的に小児剤形に対応する薬局を設置するなど、
小児剤形を利用しやすくなる仕組みを検討すべきこととした。
(4)検討会後の対応状況
検討会の議論を踏まえ、次の通知を発出した。
・「成人を対象とした医薬品の開発期間中に行う小児用医薬品の開発計画の策定につ
いて」
(令和 6 年 1 月 12 日医薬薬審発 0112 第3号厚生労働省医薬局医薬品審査管
理課長通知)
4.我が国の承認審査における日本人データの必要性の整理(国際共同治験に参加する場
合の日本人第1相試験の必要性)
(1)背景
近年、創薬環境の変化に伴い、大手製薬企業であっても創薬シーズをベンチャー等
の新興バイオ医薬品企業に依存する傾向が強まっている。欧米の新興バイオ医薬品企
業は、経営上の事情等から、開発の早期段階では、日本での開発を欧米と同時に行う
ことは少ないため、大手製薬企業が導入した以降(主に第2相試験の終了後)に日本
での開発を検討・着手するケースが増加している。
現在、国際共同治験に日本が参加するにあたって、日本人での安全性に関する説明
が十分になされない場合は、事前に日本人での第1相試験を実施する必要があるとさ
れている。一方、日本人第1相試験を実施するためには、一定の時間と費用を要する
ことから、それによって第3相試験の開始が遅延したり、それを回避するために日本
での開発を諦めるケースがあるとの指摘がある。治験における安全性の確保と、新薬