






















よむ、つかう、まなぶ。
【資料2】テーマ②(新技術による医薬品等にも対応したリスクに基づく市販後安全性対策の強化、法違反事例を踏まえた更なる法令遵守や品質確保の取組の実施)について.pdf (24 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_44832.html |
| 出典情報 | 厚生科学審議会 医薬品医療機器制度部会(令和6年度第8回 10/31)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
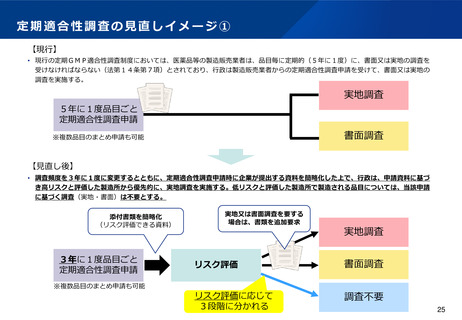
(4)適合性調査(GMP)の見直しについて
検討の方向性(案)
•
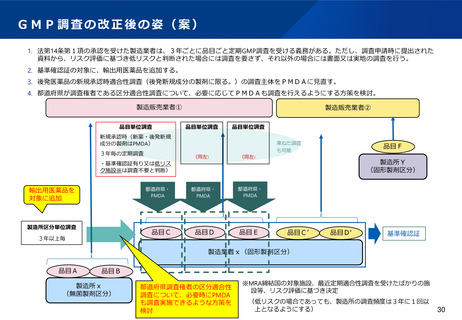
新薬や後発医薬品の新規承認時におけるGMP適合性調査については、実地調査を原則とする運用を行う。
•
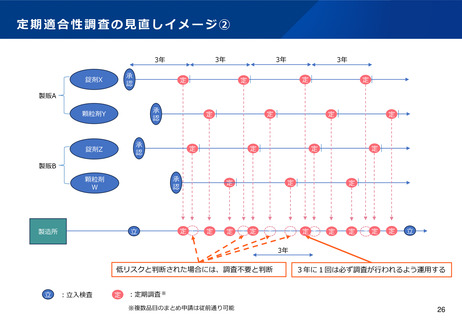
現行制度において5年に1度実施される定期のGMP適合性調査については、調査頻度を3年に1度に変更する。さらに、調
査申請時に提出される初期資料をもとにリスク評価を行い、低リスクと評価された場合は調査不要とし、品質管理上のリス
クの高い製造所(高リスク製造所)に対して確実に高頻度で実地調査を行えるようにすることで、製造所に対する監視指導
を強化することとしてはどうか。
•
上記の実地によるGMP適合性調査や、法第69条に基づく無通告の立入検査を高リスク製造所に対して重点的に実施していく
ことが重要であり、その際、どういった観点でリスクを判断していくか基準が必要。
•
具体的には、調査の対象となる品目や製造所の特性について以下のような点を踏まえてリスク評価を実施することとしては
どうか。
【品目の特性】
➢ 剤形
➢ 製品のリスク(生物学的製剤、無菌製剤等)
➢ 特殊な製造技術によるものか
➢ 滅菌・無菌操作の有無、製造工程の複雑さ
等
【製造所の特性】
➢ 当該製造所に対する過去の調査履歴(各製造所に対して3年に1度は必ず調査が行われるように運用)
➢ MRAの対象となる品目を製造する施設について、MRA締結国の施設かどうか
➢ 製造所の従業員数
➢ 製造管理・品質管理に係る不正事案が発覚した場合の影響範囲の大きさ
等
•
基準確認証制度については、輸出用医薬品に係る定期のGMP適合性調査も対象としてはどうか。
24
検討の方向性(案)
•
新薬や後発医薬品の新規承認時におけるGMP適合性調査については、実地調査を原則とする運用を行う。
•
現行制度において5年に1度実施される定期のGMP適合性調査については、調査頻度を3年に1度に変更する。さらに、調
査申請時に提出される初期資料をもとにリスク評価を行い、低リスクと評価された場合は調査不要とし、品質管理上のリス
クの高い製造所(高リスク製造所)に対して確実に高頻度で実地調査を行えるようにすることで、製造所に対する監視指導
を強化することとしてはどうか。
•
上記の実地によるGMP適合性調査や、法第69条に基づく無通告の立入検査を高リスク製造所に対して重点的に実施していく
ことが重要であり、その際、どういった観点でリスクを判断していくか基準が必要。
•
具体的には、調査の対象となる品目や製造所の特性について以下のような点を踏まえてリスク評価を実施することとしては
どうか。
【品目の特性】
➢ 剤形
➢ 製品のリスク(生物学的製剤、無菌製剤等)
➢ 特殊な製造技術によるものか
➢ 滅菌・無菌操作の有無、製造工程の複雑さ
等
【製造所の特性】
➢ 当該製造所に対する過去の調査履歴(各製造所に対して3年に1度は必ず調査が行われるように運用)
➢ MRAの対象となる品目を製造する施設について、MRA締結国の施設かどうか
➢ 製造所の従業員数
➢ 製造管理・品質管理に係る不正事案が発覚した場合の影響範囲の大きさ
等
•
基準確認証制度については、輸出用医薬品に係る定期のGMP適合性調査も対象としてはどうか。
24