






















よむ、つかう、まなぶ。
【資料2】テーマ②(新技術による医薬品等にも対応したリスクに基づく市販後安全性対策の強化、法違反事例を踏まえた更なる法令遵守や品質確保の取組の実施)について.pdf (25 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_44832.html |
| 出典情報 | 厚生科学審議会 医薬品医療機器制度部会(令和6年度第8回 10/31)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
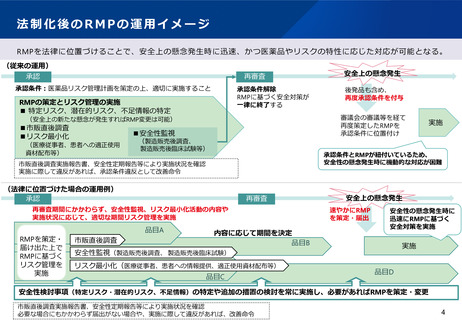
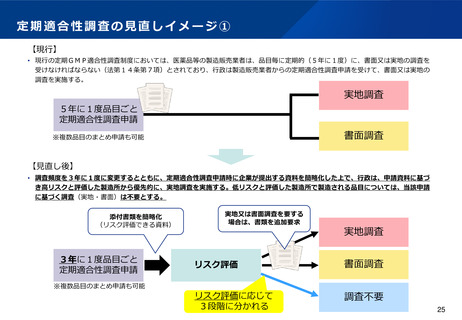
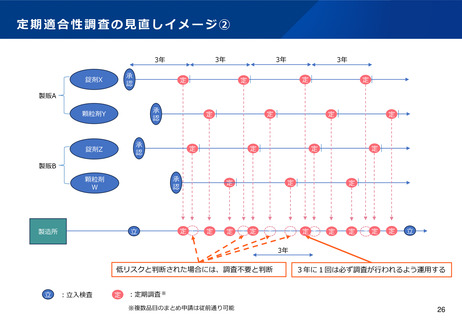
定期適合性調査の見直しイメージ①
【現行】
• 現行の定期GMP適合性調査制度においては、医薬品等の製造販売業者は、品目毎に定期的(5年に1度)に、書面又は実地の調査を
受けなければならない(法第14条第7項)とされており、行政は製造販売業者からの定期適合性調査申請を受けて、書面又は実地の
調査を実施する。
実地調査
5年に1度品目ごと
定期適合性調査申請
書面調査
※複数品目のまとめ申請も可能
【見直し後】
• 調査頻度を3年に1度に変更するとともに、定期適合性調査申請時に企業が提出する資料を簡略化した上で、行政は、申請資料に基づ
き高リスクと評価した製造所から優先的に、実地調査を実施する。低リスクと評価した製造所で製造される品目については、当該申請
に基づく調査(実地・書面)は不要とする。
添付書類を簡略化
(リスク評価できる資料)
3年に1度品目ごと
定期適合性調査申請
実地又は書面調査を要する
場合は、書類を追加要求
実地調査
リスク評価
書面調査
リスク評価に応じて
3段階に分かれる
調査不要
※複数品目のまとめ申請も可能
25
【現行】
• 現行の定期GMP適合性調査制度においては、医薬品等の製造販売業者は、品目毎に定期的(5年に1度)に、書面又は実地の調査を
受けなければならない(法第14条第7項)とされており、行政は製造販売業者からの定期適合性調査申請を受けて、書面又は実地の
調査を実施する。
実地調査
5年に1度品目ごと
定期適合性調査申請
書面調査
※複数品目のまとめ申請も可能
【見直し後】
• 調査頻度を3年に1度に変更するとともに、定期適合性調査申請時に企業が提出する資料を簡略化した上で、行政は、申請資料に基づ
き高リスクと評価した製造所から優先的に、実地調査を実施する。低リスクと評価した製造所で製造される品目については、当該申請
に基づく調査(実地・書面)は不要とする。
添付書類を簡略化
(リスク評価できる資料)
3年に1度品目ごと
定期適合性調査申請
実地又は書面調査を要する
場合は、書類を追加要求
実地調査
リスク評価
書面調査
リスク評価に応じて
3段階に分かれる
調査不要
※複数品目のまとめ申請も可能
25