











































よむ、つかう、まなぶ。
(参考資料2)業界からの要望事項.pdf (15 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_40241.html |
| 出典情報 | 厚生科学審議会 医薬品医療機器制度部会(令和6年度第2回 5/16)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
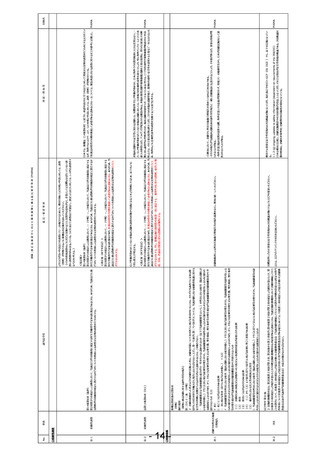
第14条第6項(抜粋)
第2項第3号の規定による審査において、当該品目が同項後段に規定する厚生労働省令で定める医薬品であるときは、あらかじめ、当該品目に係
る資料が同項後段の規定に適合するかどうかについての書面による調査又は実地の調査を行うものとする。
法第14条第6項(同上)
信頼性調査
信-2
該当法令等
信頼性調査
事項
信-1
〇信頼性調査
No.
団体名
GCP省令 第31条
2 実施医療機関の⾧は、第20条第2項及び第3項、第26条の6第2項並びに第48条第2項及び第3項の規定により通知を受けたとき、第 国際基準に合わせ重要な安全性情報のみIRB審議対象とするようにGCPを変更いただきたい。
54条第3項の規定により報告を受けたときその他実施医療機関の⾧が必要があると認めたときは、当該実施医療機関において治験を継続して行う
ことの適否について、前条第1項の規定により意見を聴いた治験審査委員会(当該治験を継続して行うことの適否の判断の前提となる特定の専門 または、GCPガイドラインでその旨を追加いただきたい。
的事項について前条第4項の規定により意見を聞いた専門治験審査委員会がある場合にあっては、同条第1項の規定により意見を聴いた治験審
査委員会及び当該専門治験審査委員会)の意見を聴かなければならない。
治-2
PhRMA
他国では重要な安全性情報のみIRB審議対象となっており、現在改訂中のICH-GCP(E6(R3))でも、以下の記載となってい
る。
1.1.2 (g) ongoing updates to safety information (dependent on requirements of the IRB/IEC)
PhRMA
日本は他国と比較し、治験実施期間中のIRB回数が2倍以上となっており、その主な理由が安全性情報の審議である。IRB審議回
数の増加は、治験の非効率化・治験費用の高額化の要因となっている。
・日欧米において、治験中に外国市販後自発報告を求めているのは日本のみである。
・それらの報告が当該臨床試験由来の副作用等報告と一緒に治験施設に伝達されることにより、その量が多くなり、重要な情報が埋
もれるリスクがある。
・海外企業と臨床開発を提携する際、海外企業にその意義が理解されず、情報フローの複雑性等から、日本が治験実施国として選
択されないリスクがある。
非臨床試験のうち安全性に関わる試験には、国際基準でGLP準拠が求められ、日本と海外でその適用範囲に大きな差異はない。一
方、GLP準拠が求められていない非臨床試験に対しては、日本独自の信頼性基準を課すことにより、特に海外ベンチャーにとって日本
<改正案(赤字を追記)>
参入の障壁となり、ドラッグラグが促進される懸念がある。非臨床薬理試験や薬物動態試験の主要な役割は、得られた結果とその解
第2項第3号の規定による審査において、・・(中略)・・。この場合において、当該品目が同項後段に規定する 釈に対し論理的な考察が行われ、臨床試験の開始をサポートすることにあると考える。ヒトでの有効性や薬物動態が確認された申請 PhRMA
厚生労働省令で定める医薬品であり、医薬品の特性その他を勘案して必要があると認めるときは、あらかじめ、当 時点では、その主要な役割は終えており、これらの非臨床成績単独で、薬剤の有効性・安全性を説明する主要なデータとはならないた
該品目に係る資料が同項後段の規定に適合するかどうかについての書面による調査又は実地の調査を行うこと め、海外のように信頼性基準の調査を免除することは妥当と考える。
ができるものとする。但し、薬機法施行規則第40条第一項に規定する二 薬理作用に関する資料、並びにホ 吸
収、分布、代謝及び排泄に関する資料は対象外とする。
GLP準拠が求められていない非臨床試験を調査の対象から免除となることをより明確にするため、以下のように
「但し書き」で明記する。
<改正案(赤字を追記)>
第2項第3号の規定による審査において、・・(中略)・・。この場合において、当該品目が同項後段に規定する
厚生労働省令で定める医薬品であり、医薬品の特性その他を勘案して必要があると認めるときは、あらかじめ、当
該品目に係る資料が同項後段の規定に適合するかどうかについての書面による調査又は実地の調査を行うこと
ができるものとする。
治-1
IRB
背景・理由等
<現記載>
第14条第6項(抜粋)
日本では、薬機法(14条第6項)に基づき、承認申請された全ての品目に対して「書面又は実地の調査を行うものとする」とされてい
第2項第3号の規定による審査において、・・(中略)・・。この場合において、当該品目が同項後段に規定する るが、海外ではリスクベースで、リスクの低い申請に対しては、調査(査察)が省略されている。
厚生労働省令で定める医薬品であるときは、あらかじめ、当該品目に係る資料が同項後段の規定に適合するか 「医薬品の特性その他を勘案して必要があると認めるときは・・」については、緊急承認における調査に関する文言を参考に記載した。 PhRMA
どうかについての書面による調査又は実地の調査を行うものとする。
よりリスクが高い申請品目の調査にリソースを配分するためにも、他国同様にリスクの低い申請に対しては、調査
(査察)が省略できるように薬機法の記載を見直していただきたい。
(HIVの書面調査のように形だけ実施することに実質的な意味がなく、企業としては実際はほぼ行っていない調
査に調査費用のみ支払っている。リスクに応じて調査品目を選定し、選定した品目に対しては、しっかりと調査をす
るべきかと考える。)
意見・要望事項
別添:改 正 法 案 提 出 に 向 け た 薬 事 規 制 に 係 る 意 見 要 望 事 項(PhRMA)
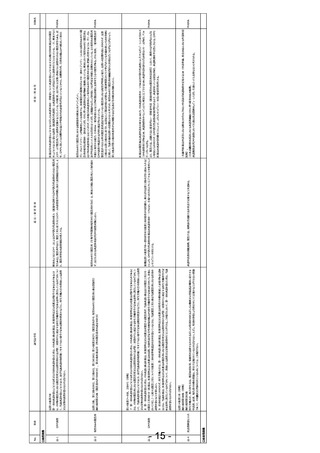
薬機法第80条の2第6項
(省略)
施行規則
(薬物に係る治験に関する副作用等の報告)
第二百七十三条 (中略)
2 治験の依頼をした者又は自ら治験を実施した者は、治験使用薬について次の各号に掲げる事項を知つたときは、それぞれ当該各号に定める期
間内にその旨を厚生労働大臣に報告しなければならない。ただし、第一号並びに第二号イ及びロについては、当該治験における被験者保護に関する
安全性の判断に影響を与えるおそれがないと認められるときは、この限りでない。
一 当該被験薬又は当該被験薬と成分が同一性を有すると認められるもの(以下「当該被験薬等」という。)の外国における使用(臨床試験にお
ける使用を除く。)で生じた次に掲げる症例等の発生のうち、当該被験薬等の副作用によるものと疑われるもの又はそれらの使用によるものと疑われ
る感染症によるものであり、かつ、そのような症例等の発生又は発生数、発生頻度、発生条件等の発生傾向が当該被験薬の治験薬概要書から予
測できないもの 七日
治験中の外国市販後
イ 死亡
治験依頼者による外国市販後自発報告等の報告義務をなくし、欧米と統一していただきたい。
自発報告
ロ 死亡につながるおそれのある症例
二 次に掲げる事項(前号に掲げるものを除く。) 十五日
イ 当該被験薬等の外国における使用(臨床試験における使用を除く。)で生じた次に掲げる症例等の発生のうち、当該被験薬等の副作用による
ものと疑われるもの又はそれらの使用によるものと疑われる感染症によるものであり、かつ、そのような症例等の発生又は発生数、発生頻度、発生条件
等の発生傾向が当該被験薬の治験薬概要書から予測できないもの
(1) 治療のために病院又は診療所への入院又は入院期間の延⾧が必要とされる症例
(2) 障害
(3) 障害につながるおそれのある症例
(4) (1)から(3)まで並びに前号イ及びロに掲げる症例に準じて重篤である症例
(5) 後世代における先天性の疾病又は異常
ロ 当該被験薬等の外国における使用(臨床試験における使用を除く。)で生じた前号イ又はロに掲げる症例等の発生のうち、当該被験薬等の副
作用によるものと疑われるもの又はそれらの使用によるものと疑われる感染症によるもの
〇治験関係
- 14 -
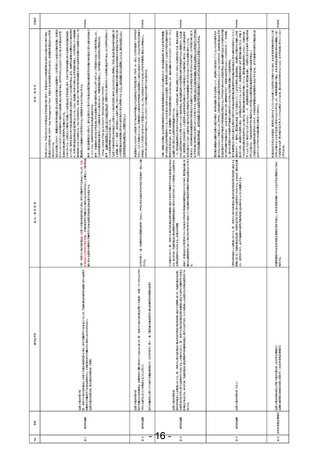
第2項第3号の規定による審査において、当該品目が同項後段に規定する厚生労働省令で定める医薬品であるときは、あらかじめ、当該品目に係
る資料が同項後段の規定に適合するかどうかについての書面による調査又は実地の調査を行うものとする。
法第14条第6項(同上)
信頼性調査
信-2
該当法令等
信頼性調査
事項
信-1
〇信頼性調査
No.
団体名
GCP省令 第31条
2 実施医療機関の⾧は、第20条第2項及び第3項、第26条の6第2項並びに第48条第2項及び第3項の規定により通知を受けたとき、第 国際基準に合わせ重要な安全性情報のみIRB審議対象とするようにGCPを変更いただきたい。
54条第3項の規定により報告を受けたときその他実施医療機関の⾧が必要があると認めたときは、当該実施医療機関において治験を継続して行う
ことの適否について、前条第1項の規定により意見を聴いた治験審査委員会(当該治験を継続して行うことの適否の判断の前提となる特定の専門 または、GCPガイドラインでその旨を追加いただきたい。
的事項について前条第4項の規定により意見を聞いた専門治験審査委員会がある場合にあっては、同条第1項の規定により意見を聴いた治験審
査委員会及び当該専門治験審査委員会)の意見を聴かなければならない。
治-2
PhRMA
他国では重要な安全性情報のみIRB審議対象となっており、現在改訂中のICH-GCP(E6(R3))でも、以下の記載となってい
る。
1.1.2 (g) ongoing updates to safety information (dependent on requirements of the IRB/IEC)
PhRMA
日本は他国と比較し、治験実施期間中のIRB回数が2倍以上となっており、その主な理由が安全性情報の審議である。IRB審議回
数の増加は、治験の非効率化・治験費用の高額化の要因となっている。
・日欧米において、治験中に外国市販後自発報告を求めているのは日本のみである。
・それらの報告が当該臨床試験由来の副作用等報告と一緒に治験施設に伝達されることにより、その量が多くなり、重要な情報が埋
もれるリスクがある。
・海外企業と臨床開発を提携する際、海外企業にその意義が理解されず、情報フローの複雑性等から、日本が治験実施国として選
択されないリスクがある。
非臨床試験のうち安全性に関わる試験には、国際基準でGLP準拠が求められ、日本と海外でその適用範囲に大きな差異はない。一
方、GLP準拠が求められていない非臨床試験に対しては、日本独自の信頼性基準を課すことにより、特に海外ベンチャーにとって日本
<改正案(赤字を追記)>
参入の障壁となり、ドラッグラグが促進される懸念がある。非臨床薬理試験や薬物動態試験の主要な役割は、得られた結果とその解
第2項第3号の規定による審査において、・・(中略)・・。この場合において、当該品目が同項後段に規定する 釈に対し論理的な考察が行われ、臨床試験の開始をサポートすることにあると考える。ヒトでの有効性や薬物動態が確認された申請 PhRMA
厚生労働省令で定める医薬品であり、医薬品の特性その他を勘案して必要があると認めるときは、あらかじめ、当 時点では、その主要な役割は終えており、これらの非臨床成績単独で、薬剤の有効性・安全性を説明する主要なデータとはならないた
該品目に係る資料が同項後段の規定に適合するかどうかについての書面による調査又は実地の調査を行うこと め、海外のように信頼性基準の調査を免除することは妥当と考える。
ができるものとする。但し、薬機法施行規則第40条第一項に規定する二 薬理作用に関する資料、並びにホ 吸
収、分布、代謝及び排泄に関する資料は対象外とする。
GLP準拠が求められていない非臨床試験を調査の対象から免除となることをより明確にするため、以下のように
「但し書き」で明記する。
<改正案(赤字を追記)>
第2項第3号の規定による審査において、・・(中略)・・。この場合において、当該品目が同項後段に規定する
厚生労働省令で定める医薬品であり、医薬品の特性その他を勘案して必要があると認めるときは、あらかじめ、当
該品目に係る資料が同項後段の規定に適合するかどうかについての書面による調査又は実地の調査を行うこと
ができるものとする。
治-1
IRB
背景・理由等
<現記載>
第14条第6項(抜粋)
日本では、薬機法(14条第6項)に基づき、承認申請された全ての品目に対して「書面又は実地の調査を行うものとする」とされてい
第2項第3号の規定による審査において、・・(中略)・・。この場合において、当該品目が同項後段に規定する るが、海外ではリスクベースで、リスクの低い申請に対しては、調査(査察)が省略されている。
厚生労働省令で定める医薬品であるときは、あらかじめ、当該品目に係る資料が同項後段の規定に適合するか 「医薬品の特性その他を勘案して必要があると認めるときは・・」については、緊急承認における調査に関する文言を参考に記載した。 PhRMA
どうかについての書面による調査又は実地の調査を行うものとする。
よりリスクが高い申請品目の調査にリソースを配分するためにも、他国同様にリスクの低い申請に対しては、調査
(査察)が省略できるように薬機法の記載を見直していただきたい。
(HIVの書面調査のように形だけ実施することに実質的な意味がなく、企業としては実際はほぼ行っていない調
査に調査費用のみ支払っている。リスクに応じて調査品目を選定し、選定した品目に対しては、しっかりと調査をす
るべきかと考える。)
意見・要望事項
別添:改 正 法 案 提 出 に 向 け た 薬 事 規 制 に 係 る 意 見 要 望 事 項(PhRMA)
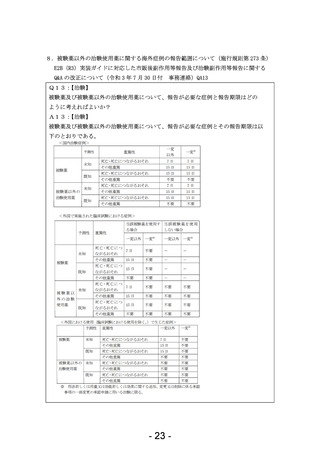
薬機法第80条の2第6項
(省略)
施行規則
(薬物に係る治験に関する副作用等の報告)
第二百七十三条 (中略)
2 治験の依頼をした者又は自ら治験を実施した者は、治験使用薬について次の各号に掲げる事項を知つたときは、それぞれ当該各号に定める期
間内にその旨を厚生労働大臣に報告しなければならない。ただし、第一号並びに第二号イ及びロについては、当該治験における被験者保護に関する
安全性の判断に影響を与えるおそれがないと認められるときは、この限りでない。
一 当該被験薬又は当該被験薬と成分が同一性を有すると認められるもの(以下「当該被験薬等」という。)の外国における使用(臨床試験にお
ける使用を除く。)で生じた次に掲げる症例等の発生のうち、当該被験薬等の副作用によるものと疑われるもの又はそれらの使用によるものと疑われ
る感染症によるものであり、かつ、そのような症例等の発生又は発生数、発生頻度、発生条件等の発生傾向が当該被験薬の治験薬概要書から予
測できないもの 七日
治験中の外国市販後
イ 死亡
治験依頼者による外国市販後自発報告等の報告義務をなくし、欧米と統一していただきたい。
自発報告
ロ 死亡につながるおそれのある症例
二 次に掲げる事項(前号に掲げるものを除く。) 十五日
イ 当該被験薬等の外国における使用(臨床試験における使用を除く。)で生じた次に掲げる症例等の発生のうち、当該被験薬等の副作用による
ものと疑われるもの又はそれらの使用によるものと疑われる感染症によるものであり、かつ、そのような症例等の発生又は発生数、発生頻度、発生条件
等の発生傾向が当該被験薬の治験薬概要書から予測できないもの
(1) 治療のために病院又は診療所への入院又は入院期間の延⾧が必要とされる症例
(2) 障害
(3) 障害につながるおそれのある症例
(4) (1)から(3)まで並びに前号イ及びロに掲げる症例に準じて重篤である症例
(5) 後世代における先天性の疾病又は異常
ロ 当該被験薬等の外国における使用(臨床試験における使用を除く。)で生じた前号イ又はロに掲げる症例等の発生のうち、当該被験薬等の副
作用によるものと疑われるもの又はそれらの使用によるものと疑われる感染症によるもの
〇治験関係
- 14 -