















































よむ、つかう、まなぶ。
【資料1-1】 医薬品等行政評価・監視委員会における海外調査の状況 (14 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_24538.html |
| 出典情報 | 医薬品等行政評価・監視委員会(第7回 3/18)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
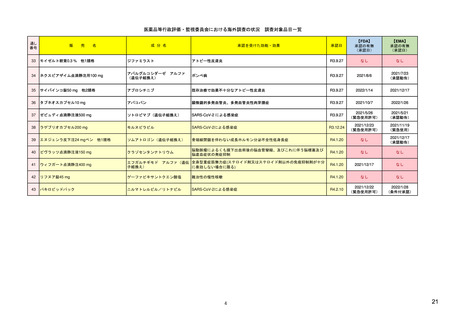
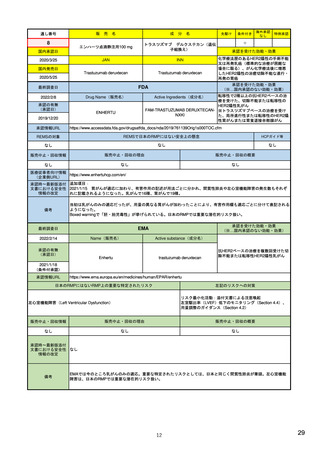
ア.別添「市販後における医薬品の副作用情報収集及び評価のシステム」
R4.3.4
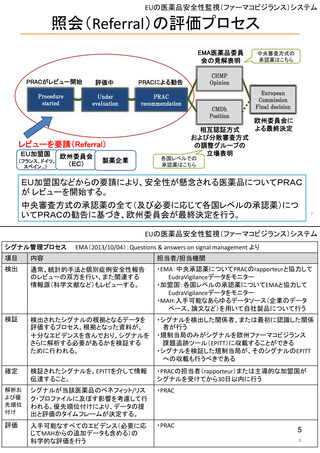
米国
欧州
日本
1)市販後の副作用報告(主に製薬企業からの個別症例安全性報告)
ICH E2Bフォーマット
ICH E2Bガイドライン
ICH E2Bフォーマット
ICH E2Bガイドライン、EU-
ICH E2Bフォーマット
GVP module VI
報告フォーマット
ICH E2Bガイドライン
21)https://www.jshp.or.jp/co
nt/21/0805-5-1.pdf
米国で承認されている医薬品
(国内承認薬と同一の有効成分に関する外国
での有害事象報告を含む)
1)
https://www.fda.gov/files/vac
cines%2C%20blood%20%26%
20biologics/published/DraftGuidance-for-Industry--
報告対象医薬品
欧州経済地域(EEA)で認可された医薬品
10 ) Guideline on good
日本国内で承認されている医薬品
(中央審査方式、非中央審査方式)
pharmacovigilance practices
(国内承認薬と同一の有効成分に関する外国での ourei/doc/tsuchi/T210802I00
(EEA域内承認薬と同一の有効成分に関する
EEA域外での副作用報告を含む)
(GVP) - Module VI – Collection,
management and submission of
副作用報告を含む)
22)https://www.mhlw.go.jp/h
60.pdf (p.4 Q&A8)
reports of suspected adverse
Postmarketing-Safety-
reactions to medicinal products
Reporting-for-Human-Drug-
(Rev 2) (europa.eu)
and-Biological-ProductsIncluding-Vaccines.pdf
・未知・重篤の有害事象。
1)同上 (202-294行目,
・医薬品と有害事象の間に合理的な因果関
・ただし、市販後試験からの有害事象は、
Appedix A,)
係の可能性がある副作用(自発報告は因果
ない症例であって、重篤な症例。軽微でなく、
関係不明でも報告対象となる)
かつ、添付文書等から予測できない未知の副作
医薬品との間に合理的な因果関係の可能性
報告対象副作用
10) (p.6 VI.A.1.1.)
・医薬品等との因果関係が(完全に)否定でき 22)(p.3 Q&A1)
があると考えるもののみ個別報告の対象。
用。
・取下げの制度は無し。
・非重篤であることが判明又は因果関係がない
・複数症例を含む文献情報は、症例ごとに報
と判断された場合、報告対象外となる。
告する。
・未知・重篤の有害事象:15日以内
報告期限
1) (350-422行目)
・全ての重篤な疑わしい副作用:15日以内
10) (p.40 VI.C.3.)
・未知の副作用(外国の症例も含む)、死亡、 22) (p.8 Q&A16-17)
(既知・重篤、既知および未知・非重篤は、
・全ての非重篤な疑わしい副作用:90日以
承認後2年以内の医薬品の既知・重篤副作用:
定期安全性報告として提出)
内
15日以内
・承認後2年以上の医薬品の既知・重篤副作
用:30日以内
既知の重篤有害事象も、上記の”15日報告”と 1)(598-601行目)
・重複報告を回避し、重複が判明した場合
11)
重複が判明した場合には、取り下げ報告とす
23)
して提出可能であり、その場合、重複を避け
には統合処理する。
https://www.ema.europa.eu/en
る。
https://www.pmda.go.jp/files
るため定期安全性報告には含めない。
・文献情報については、医学文献モニタリ
症例に関する重複報告の扱い
/documents/regulatory-
/000236488.pdf(p29 1(1))
procedural-guideline/guideline-
ングリストの対象薬に関して、EMAが重複
good-pharmacovigilance-
チェックを行う(リスト対象外の成分に関
practices-gvp-module-vi-
する文献情報は、企業が報告するものとな
addendum-i-duplicate-
り、重複があり得る)。
management-suspected_en.pdf
12)https://www.ema.europa.eu/
en/human-regulatory/postauthorisation/pharmacovigilance
/medical-literature-monitoring
有効な症例報告の条件
(1)識別可能な報告者、(2)識別可能な患者、 1)(296-307行目)
(1)識別可能な報告者、(2)識別可能な患者、 10) (p14, VI.B2.)
(1)識別可能な報告者、(2)識別可能な患者、(3) 21) p4
(3)被疑薬、(4)有害事象
(3)被疑薬、(4)副作用
被疑薬、(4)副作用
(上記4条件が揃えばインターネット上で得
た有害事象情報も報告する)
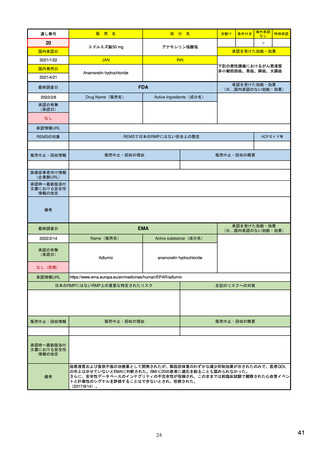
14
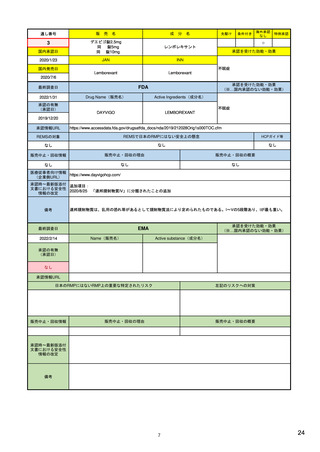
R4.3.4
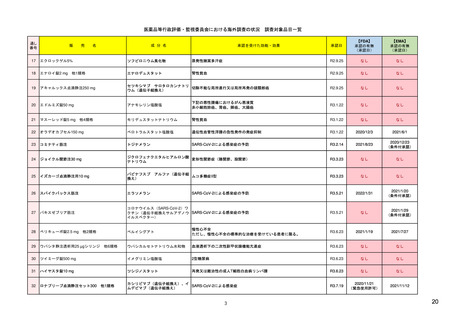
米国
欧州
日本
1)市販後の副作用報告(主に製薬企業からの個別症例安全性報告)
ICH E2Bフォーマット
ICH E2Bガイドライン
ICH E2Bフォーマット
ICH E2Bガイドライン、EU-
ICH E2Bフォーマット
GVP module VI
報告フォーマット
ICH E2Bガイドライン
21)https://www.jshp.or.jp/co
nt/21/0805-5-1.pdf
米国で承認されている医薬品
(国内承認薬と同一の有効成分に関する外国
での有害事象報告を含む)
1)
https://www.fda.gov/files/vac
cines%2C%20blood%20%26%
20biologics/published/DraftGuidance-for-Industry--
報告対象医薬品
欧州経済地域(EEA)で認可された医薬品
10 ) Guideline on good
日本国内で承認されている医薬品
(中央審査方式、非中央審査方式)
pharmacovigilance practices
(国内承認薬と同一の有効成分に関する外国での ourei/doc/tsuchi/T210802I00
(EEA域内承認薬と同一の有効成分に関する
EEA域外での副作用報告を含む)
(GVP) - Module VI – Collection,
management and submission of
副作用報告を含む)
22)https://www.mhlw.go.jp/h
60.pdf (p.4 Q&A8)
reports of suspected adverse
Postmarketing-Safety-
reactions to medicinal products
Reporting-for-Human-Drug-
(Rev 2) (europa.eu)
and-Biological-ProductsIncluding-Vaccines.pdf
・未知・重篤の有害事象。
1)同上 (202-294行目,
・医薬品と有害事象の間に合理的な因果関
・ただし、市販後試験からの有害事象は、
Appedix A,)
係の可能性がある副作用(自発報告は因果
ない症例であって、重篤な症例。軽微でなく、
関係不明でも報告対象となる)
かつ、添付文書等から予測できない未知の副作
医薬品との間に合理的な因果関係の可能性
報告対象副作用
10) (p.6 VI.A.1.1.)
・医薬品等との因果関係が(完全に)否定でき 22)(p.3 Q&A1)
があると考えるもののみ個別報告の対象。
用。
・取下げの制度は無し。
・非重篤であることが判明又は因果関係がない
・複数症例を含む文献情報は、症例ごとに報
と判断された場合、報告対象外となる。
告する。
・未知・重篤の有害事象:15日以内
報告期限
1) (350-422行目)
・全ての重篤な疑わしい副作用:15日以内
10) (p.40 VI.C.3.)
・未知の副作用(外国の症例も含む)、死亡、 22) (p.8 Q&A16-17)
(既知・重篤、既知および未知・非重篤は、
・全ての非重篤な疑わしい副作用:90日以
承認後2年以内の医薬品の既知・重篤副作用:
定期安全性報告として提出)
内
15日以内
・承認後2年以上の医薬品の既知・重篤副作
用:30日以内
既知の重篤有害事象も、上記の”15日報告”と 1)(598-601行目)
・重複報告を回避し、重複が判明した場合
11)
重複が判明した場合には、取り下げ報告とす
23)
して提出可能であり、その場合、重複を避け
には統合処理する。
https://www.ema.europa.eu/en
る。
https://www.pmda.go.jp/files
るため定期安全性報告には含めない。
・文献情報については、医学文献モニタリ
症例に関する重複報告の扱い
/documents/regulatory-
/000236488.pdf(p29 1(1))
procedural-guideline/guideline-
ングリストの対象薬に関して、EMAが重複
good-pharmacovigilance-
チェックを行う(リスト対象外の成分に関
practices-gvp-module-vi-
する文献情報は、企業が報告するものとな
addendum-i-duplicate-
り、重複があり得る)。
management-suspected_en.pdf
12)https://www.ema.europa.eu/
en/human-regulatory/postauthorisation/pharmacovigilance
/medical-literature-monitoring
有効な症例報告の条件
(1)識別可能な報告者、(2)識別可能な患者、 1)(296-307行目)
(1)識別可能な報告者、(2)識別可能な患者、 10) (p14, VI.B2.)
(1)識別可能な報告者、(2)識別可能な患者、(3) 21) p4
(3)被疑薬、(4)有害事象
(3)被疑薬、(4)副作用
被疑薬、(4)副作用
(上記4条件が揃えばインターネット上で得
た有害事象情報も報告する)
14