



























よむ、つかう、まなぶ。
【資料1】テーマ①(ドラッグロスや供給不足などの医薬品等へのアクセスの課題に対応した安全かつ迅速な承認制度の確立)について.pdf (28 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_40580.html |
| 出典情報 | 厚生科学審議会 医薬品医療機器制度部会(令和6年度第3回 6/6)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
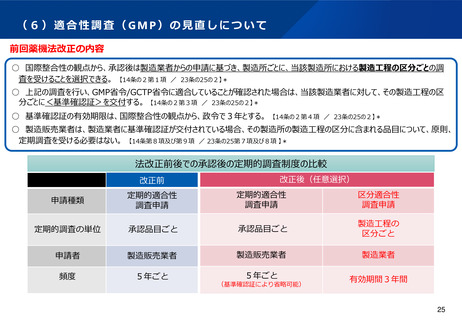
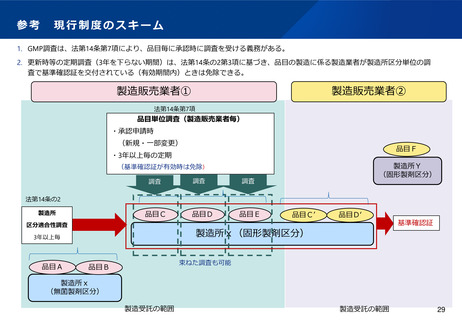
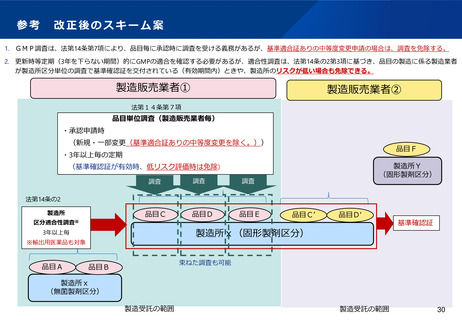
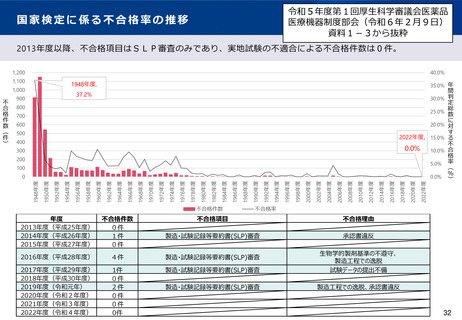
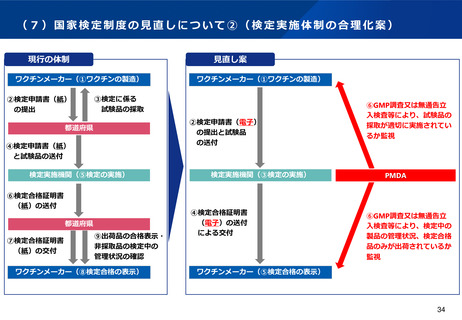
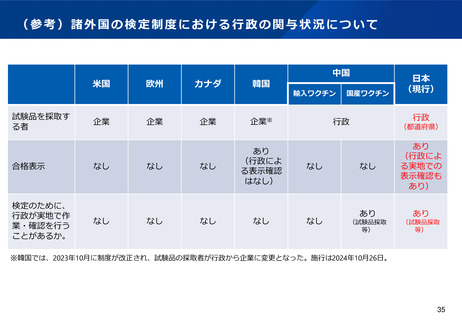
適合性調査(GMP)制度の見直しのポイント(案)
GMP適合性調査の課題
(より合理的なGMP調査体制の構築)
•
製造業者におけるGMP適合性や法令遵守の確保や取締強化が、近年の薬機法違反事例からみて課題である。
•
法第14条第7項のGMPの適合性調査は、すべての対象品目について、「書面又は実地」の調査を行うこととされているが、調
査・査察のリソースに限りがあるため、書面調査で対応するケースが、PMDAでは令和5年度の実績で約87%を占めている。特
に、新薬・後発の新規品目の承認では、品質リスクが発生しやすく、本来実地調査に注力できるリソースを確保したい。
•
薬機法違反事案が増加する中、PMDAや都道府県は、違反が疑われる製造所や不適合リスクが高い製造所に対し、重点的に法第
69条による無通告「立入検査」を実施している。このような「立入検査」や実地の法第14条第7項のGMP調査をリスクの高い製
造所に対して重点的に実施できるよう査察リソースを集約・拡充したい。
(基準確認証制度の合理化)
•
前回の法改正において国内流通品に係る定期のGMP調査については基準確認証制度が導入されたが、一方で輸出用医薬品は基準
確認証制度の対象外であることから、GMP調査の合理化が進んでおらず、また、そのためPMDA・都道府県のリソースの有効活
用も図られていないとの指摘がある。
(検討中の製造・品質に係る中等度変更の承認申請に係るGMP調査)
•
製造・品質に係る中等度変更の承認申請に係るGMP調査を品目個別に実施するべきかの検討が必要である。
改正の方向性
•
新薬や後発医薬品の新規品目に係る適合性調査については、実地調査を原則とする調査運用を行う。
•
一方、品目毎の定期GMP調査は、GMP適合状況を製造業者に申告させ、製造所の品質リスクや直近調査からの経過期間等を施設
毎に評価し、リスクが高い製造業者に対して集約的に実地調査を実施する制度に切り換える。
※
低リスクと評価された場合には調査を免除。製造所の評価のためには、品目毎よりも、区分適合性申請に基づく施設単位での「基準確認証」方式による情
報収集が有益である。
•
基準確認証制度については、輸出用医薬品に係る定期のGMP調査も対象とすることで、施設単位でのGMP調査の更なる合理化を
図り、PMDA・都道府県のリソースの有効活用を進めることとしてはどうか。
•
中等度の承認事項の変更(製造方法・規格及び試験方法)については、変更前の製品と品質の同等性・同質性が確認できるもの
については、施設単位での「基準確認証」が有効である範囲においては、個別のGMP適合性調査は不要としてはどうか。 28
GMP適合性調査の課題
(より合理的なGMP調査体制の構築)
•
製造業者におけるGMP適合性や法令遵守の確保や取締強化が、近年の薬機法違反事例からみて課題である。
•
法第14条第7項のGMPの適合性調査は、すべての対象品目について、「書面又は実地」の調査を行うこととされているが、調
査・査察のリソースに限りがあるため、書面調査で対応するケースが、PMDAでは令和5年度の実績で約87%を占めている。特
に、新薬・後発の新規品目の承認では、品質リスクが発生しやすく、本来実地調査に注力できるリソースを確保したい。
•
薬機法違反事案が増加する中、PMDAや都道府県は、違反が疑われる製造所や不適合リスクが高い製造所に対し、重点的に法第
69条による無通告「立入検査」を実施している。このような「立入検査」や実地の法第14条第7項のGMP調査をリスクの高い製
造所に対して重点的に実施できるよう査察リソースを集約・拡充したい。
(基準確認証制度の合理化)
•
前回の法改正において国内流通品に係る定期のGMP調査については基準確認証制度が導入されたが、一方で輸出用医薬品は基準
確認証制度の対象外であることから、GMP調査の合理化が進んでおらず、また、そのためPMDA・都道府県のリソースの有効活
用も図られていないとの指摘がある。
(検討中の製造・品質に係る中等度変更の承認申請に係るGMP調査)
•
製造・品質に係る中等度変更の承認申請に係るGMP調査を品目個別に実施するべきかの検討が必要である。
改正の方向性
•
新薬や後発医薬品の新規品目に係る適合性調査については、実地調査を原則とする調査運用を行う。
•
一方、品目毎の定期GMP調査は、GMP適合状況を製造業者に申告させ、製造所の品質リスクや直近調査からの経過期間等を施設
毎に評価し、リスクが高い製造業者に対して集約的に実地調査を実施する制度に切り換える。
※
低リスクと評価された場合には調査を免除。製造所の評価のためには、品目毎よりも、区分適合性申請に基づく施設単位での「基準確認証」方式による情
報収集が有益である。
•
基準確認証制度については、輸出用医薬品に係る定期のGMP調査も対象とすることで、施設単位でのGMP調査の更なる合理化を
図り、PMDA・都道府県のリソースの有効活用を進めることとしてはどうか。
•
中等度の承認事項の変更(製造方法・規格及び試験方法)については、変更前の製品と品質の同等性・同質性が確認できるもの
については、施設単位での「基準確認証」が有効である範囲においては、個別のGMP適合性調査は不要としてはどうか。 28