


































よむ、つかう、まなぶ。
【資料3】日米欧における医薬品の品質管理・製造管理に関する調査の状況(令和4年度欧米の薬事制度に関する調査・整理業務 調査結果) (21 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_30030.html |
| 出典情報 | 医薬品等行政評価・監視委員会(第10回 12/27)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
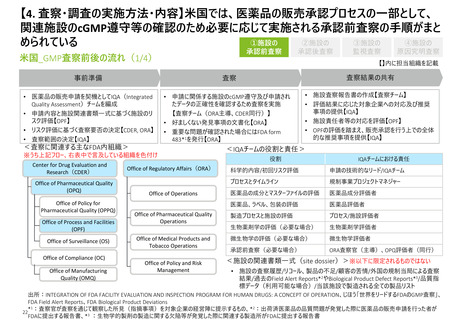
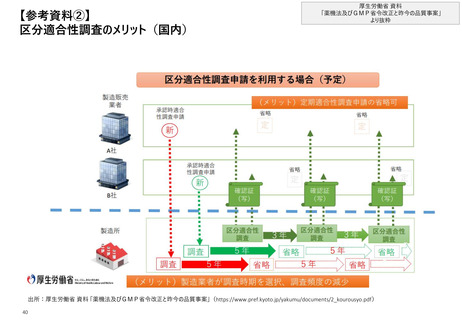
【4.査察・調査の実施方法・内容】日本では、GMP調査要領において適合性調査及び立
入検査等を包含した調査手順が示されている
①適合性調査・
確認
日本_GMP調査前後の流れ
(初回は原則2日以上)
原則調査終了日から
10業務日以内
受領後速やかに確認
確認後速やかに作成
実地調査
指摘事項を
製造所に対し提示
製造所から提出された
改善事項・改善計画の確
認、適合状況の判断
調査結果報告書作成、
写しの交付、
通知書等の送付
事前準備
•
•
•
•
基本方針の策定
査察チームの編成
調査計画の策定
事前通知*1
•
•
•
•
調査通知書の提示
調査基本事項の確認
調査実施
講評
<GMP調査の対象のあり方>
分類
承認前
適合性
調査
適
合
性
調
査
立
入
検
査
等
調査対象
• GMP適合性評価基準に
従って、不備事項を分類
• GMP調査指摘事項書を作
成
• GMP調査指摘事項書を、
調査対象製造業者等の責
任者に交付
承認申請に係る品目(製品)。但し当該製
造所としての初回調査の場合は製造所全体
初回は適合性調査申請に係る品目(製品)、
又は適合性調査を受けなければならない品目
(製品)をまとめての製造所全体。2回目以
降は特に前回調査以降変更のあった部分
初回は適合性調査申請に係る製造工程の区
区分
分に含まれる全ての品目(製品)をまとめての
適合性
製造所全体。2回目以降は特に前回調査以
調査
降変更のあった部分
変更計画に 変更計画に係る品目(製品)。但し当該製
係る適合性 造所 として初回の調査である場合においては製
確認
造所全体
承認後等
適合性
調査*2
通常検査
特別検査
②立入検査等
<不備事項の分類>
分類
定義
重度
GMP省令に抵触して
おり明白なリスクや虚
偽等が認められた
中程度
GMP省令に抵触して
いるが重度ではない
軽度
GMP省令への抵触は
明らかではないが改
善が必要
初回は製造所全体、2回目以降は製造所全
体で特に前回調査以降変更等のあった部分
調査目的により異なる
• 製造所が作成した、
改善計画書、又は改善結
果報告書の内容を確認
• 改善が確認された場合には、
監視指導措置等を行う関
係部門に連絡
• 改善内容が適切でない場
合には、是正を指導
• 調査部門の長が、適合性
評価基準に基づき適合状
況の判定を実施
仮に不適合の場合は、「薬
事監視指導要領」に従って
措置を実行
• 調査実施責任者が、
調査結果報告書を作成し、
その写しを調査対象製造
業者等に交付
• 調査チームにて、
調査結果報告書及び
関連記録を適切に保管
• 適合性調査・確認の場合
品目の製造販売業者等に
調査結果を通知
• GMP調査の実施状況につ
いて、厚生労働省に定期
的に報告
※GMP適合性評価基準を踏
まえた適合/不適合は以下の
通り
適合:不備事項なし、又は
不備事項が軽度であり、改善
結果報告書もしくは改善計画
書が確認された場合
不適合:不備事項が中程度
又は重度であり、不備事項の
分類に応じた期間内に改善が
完了しなかった場合
出所:厚生労働省 GMP 調査要領の制定について、じほう「リスクベースによるGMP監査実施ノウハウ 第2版」
*1:立入検査等の場合は原則無通告
21 2
* :「輸出用医薬品等」、「既存定期」の2つにさらに分類される。「輸出用医薬品等」の場合初回で製造所全体を対象とするのは当該製造所としての初回調査の場合のみ
入検査等を包含した調査手順が示されている
①適合性調査・
確認
日本_GMP調査前後の流れ
(初回は原則2日以上)
原則調査終了日から
10業務日以内
受領後速やかに確認
確認後速やかに作成
実地調査
指摘事項を
製造所に対し提示
製造所から提出された
改善事項・改善計画の確
認、適合状況の判断
調査結果報告書作成、
写しの交付、
通知書等の送付
事前準備
•
•
•
•
基本方針の策定
査察チームの編成
調査計画の策定
事前通知*1
•
•
•
•
調査通知書の提示
調査基本事項の確認
調査実施
講評
<GMP調査の対象のあり方>
分類
承認前
適合性
調査
適
合
性
調
査
立
入
検
査
等
調査対象
• GMP適合性評価基準に
従って、不備事項を分類
• GMP調査指摘事項書を作
成
• GMP調査指摘事項書を、
調査対象製造業者等の責
任者に交付
承認申請に係る品目(製品)。但し当該製
造所としての初回調査の場合は製造所全体
初回は適合性調査申請に係る品目(製品)、
又は適合性調査を受けなければならない品目
(製品)をまとめての製造所全体。2回目以
降は特に前回調査以降変更のあった部分
初回は適合性調査申請に係る製造工程の区
区分
分に含まれる全ての品目(製品)をまとめての
適合性
製造所全体。2回目以降は特に前回調査以
調査
降変更のあった部分
変更計画に 変更計画に係る品目(製品)。但し当該製
係る適合性 造所 として初回の調査である場合においては製
確認
造所全体
承認後等
適合性
調査*2
通常検査
特別検査
②立入検査等
<不備事項の分類>
分類
定義
重度
GMP省令に抵触して
おり明白なリスクや虚
偽等が認められた
中程度
GMP省令に抵触して
いるが重度ではない
軽度
GMP省令への抵触は
明らかではないが改
善が必要
初回は製造所全体、2回目以降は製造所全
体で特に前回調査以降変更等のあった部分
調査目的により異なる
• 製造所が作成した、
改善計画書、又は改善結
果報告書の内容を確認
• 改善が確認された場合には、
監視指導措置等を行う関
係部門に連絡
• 改善内容が適切でない場
合には、是正を指導
• 調査部門の長が、適合性
評価基準に基づき適合状
況の判定を実施
仮に不適合の場合は、「薬
事監視指導要領」に従って
措置を実行
• 調査実施責任者が、
調査結果報告書を作成し、
その写しを調査対象製造
業者等に交付
• 調査チームにて、
調査結果報告書及び
関連記録を適切に保管
• 適合性調査・確認の場合
品目の製造販売業者等に
調査結果を通知
• GMP調査の実施状況につ
いて、厚生労働省に定期
的に報告
※GMP適合性評価基準を踏
まえた適合/不適合は以下の
通り
適合:不備事項なし、又は
不備事項が軽度であり、改善
結果報告書もしくは改善計画
書が確認された場合
不適合:不備事項が中程度
又は重度であり、不備事項の
分類に応じた期間内に改善が
完了しなかった場合
出所:厚生労働省 GMP 調査要領の制定について、じほう「リスクベースによるGMP監査実施ノウハウ 第2版」
*1:立入検査等の場合は原則無通告
21 2
* :「輸出用医薬品等」、「既存定期」の2つにさらに分類される。「輸出用医薬品等」の場合初回で製造所全体を対象とするのは当該製造所としての初回調査の場合のみ