


































よむ、つかう、まなぶ。
【資料3】日米欧における医薬品の品質管理・製造管理に関する調査の状況(令和4年度欧米の薬事制度に関する調査・整理業務 調査結果) (22 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_30030.html |
| 出典情報 | 医薬品等行政評価・監視委員会(第10回 12/27)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
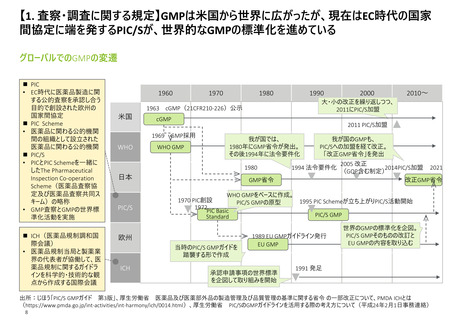
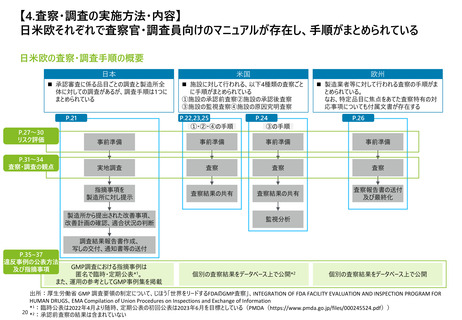
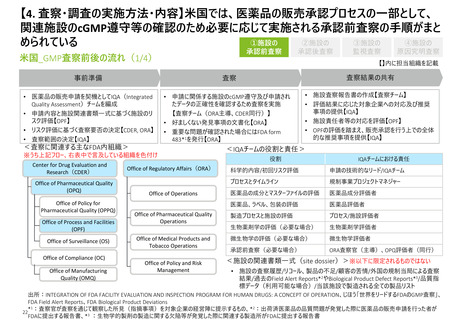
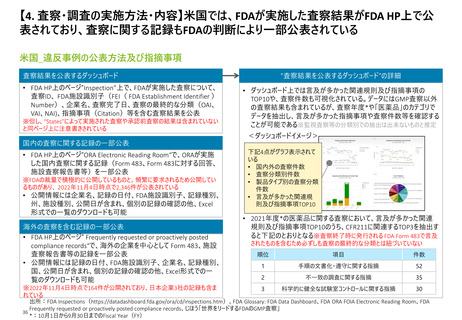
【4. 査察・調査の実施方法・内容】米国では、医薬品の販売承認プロセスの一部として、
関連施設のcGMP遵守等の確認のため必要に応じて実施される承認前査察の手順がまと
①施設の
②施設の
③施設の
④施設の
められている
承認前査察
米国_GMP査察前後の流れ(1/4)
• 申請に関係する施設のcGMP遵守及び申請され
たデータの正確性を確認するため査察を実施
【査察チーム(ORA主導、CDER同行)】
• 好ましくない発見事項の文書化【ORA】
• 重要な問題が確認された場合にはFDA form
483*1を発行【ORA】
<査察に関連する主なFDA内組織>
Office of Policy for
Pharmaceutical Quality (OPPQ)
Office of Process and Facilities
(OPF)
Office of Surveillance (OS)
Office of Compliance (OC)
Office of Manufacturing
Quality (OMQ)
• 施設査察報告書の作成【査察チーム】
• 評価結果に応じた対象企業への対応及び推奨
事項の提供【IQA】
• 施設責任者等の対応を評価【OPF】
• OPFの評価を踏まえ、販売承認を行う上での全体
的な推奨事項を提供【IQA】
<IQAチームの役割と責任>
※うち上記フロー、右表中で言及している組織を色付け
Office of Pharmaceutical Quality
(OPQ)
原因究明査察
査察結果の共有
査察
• 医薬品の販売申請を契機としてIQA(Integrated
Quality Assessment)チームを編成
• 申請内容と施設関連書類一式に基づく施設のリ
スク評価【OPF】
• リスク評価に基づく査察要否の決定【CDER, ORA】
• 査察範囲の決定【IQA】
監視査察
【】内に担当組織を記載
事前準備
Center for Drug Evaluation and
Research(CDER)
承認後査察
Office of Regulatory Affairs(ORA)
Office of Operations
Office of Pharmaceutical Quality
Operations
Office of Medical Products and
Tobacco Operations
Office of Policy and Risk
Management
役割
IQAチームにおける責任
科学的内容/初回リスク評価
申請の技術的なリード/IQAチーム
プロセスとタイムライン
規制事業プロジェクトマネジャー
医薬品の成分とマスターファイルの評価
医薬品成分評価者
医薬品、ラベル、包装の評価
医薬品評価者
製造プロセスと施設の評価
プロセス/施設評価者
生物薬剤学の評価(必要な場合)
生物薬剤学評価者
微生物学の評価(必要な場合)
微生物学評価者
承認前査察(必要な場合)
ORA査察官(主導)、OPQ評価者(同行)
<施設の関連書類一式(site dossier)>※以下に限定されるものではない
• 施設の査察履歴/リコール、製品の不足/顧客の苦情/外国の規制当局による査察
結果/過去のField Alert Reports*2やBiological Product Defect Reports*3/品質指
標データ(利用可能な場合)/当該施設で製造される全ての製品リスト
出所:INTEGRATION OF FDA FACILITY EVALUATION AND INSPECTION PROGRAM FOR HUMAN DRUGS: A CONCEPT OF OPERATION、じほう「世界をリードするFDAのGMP査察」、
FDA Field Alert Reports、FDA Biological Product Deviations
*1:査察官が査察を通じて観察した所見(指摘事項)を対象企業の経営陣に提示するもの、*2:出荷済医薬品の品質問題が発覚した際に医薬品の販売申請を行った者が
22
FDAに提出する報告書、*3 :生物学的製剤の製造に関する欠陥等が発覚した際に関連する製造所がFDAに提出する報告書
関連施設のcGMP遵守等の確認のため必要に応じて実施される承認前査察の手順がまと
①施設の
②施設の
③施設の
④施設の
められている
承認前査察
米国_GMP査察前後の流れ(1/4)
• 申請に関係する施設のcGMP遵守及び申請され
たデータの正確性を確認するため査察を実施
【査察チーム(ORA主導、CDER同行)】
• 好ましくない発見事項の文書化【ORA】
• 重要な問題が確認された場合にはFDA form
483*1を発行【ORA】
<査察に関連する主なFDA内組織>
Office of Policy for
Pharmaceutical Quality (OPPQ)
Office of Process and Facilities
(OPF)
Office of Surveillance (OS)
Office of Compliance (OC)
Office of Manufacturing
Quality (OMQ)
• 施設査察報告書の作成【査察チーム】
• 評価結果に応じた対象企業への対応及び推奨
事項の提供【IQA】
• 施設責任者等の対応を評価【OPF】
• OPFの評価を踏まえ、販売承認を行う上での全体
的な推奨事項を提供【IQA】
<IQAチームの役割と責任>
※うち上記フロー、右表中で言及している組織を色付け
Office of Pharmaceutical Quality
(OPQ)
原因究明査察
査察結果の共有
査察
• 医薬品の販売申請を契機としてIQA(Integrated
Quality Assessment)チームを編成
• 申請内容と施設関連書類一式に基づく施設のリ
スク評価【OPF】
• リスク評価に基づく査察要否の決定【CDER, ORA】
• 査察範囲の決定【IQA】
監視査察
【】内に担当組織を記載
事前準備
Center for Drug Evaluation and
Research(CDER)
承認後査察
Office of Regulatory Affairs(ORA)
Office of Operations
Office of Pharmaceutical Quality
Operations
Office of Medical Products and
Tobacco Operations
Office of Policy and Risk
Management
役割
IQAチームにおける責任
科学的内容/初回リスク評価
申請の技術的なリード/IQAチーム
プロセスとタイムライン
規制事業プロジェクトマネジャー
医薬品の成分とマスターファイルの評価
医薬品成分評価者
医薬品、ラベル、包装の評価
医薬品評価者
製造プロセスと施設の評価
プロセス/施設評価者
生物薬剤学の評価(必要な場合)
生物薬剤学評価者
微生物学の評価(必要な場合)
微生物学評価者
承認前査察(必要な場合)
ORA査察官(主導)、OPQ評価者(同行)
<施設の関連書類一式(site dossier)>※以下に限定されるものではない
• 施設の査察履歴/リコール、製品の不足/顧客の苦情/外国の規制当局による査察
結果/過去のField Alert Reports*2やBiological Product Defect Reports*3/品質指
標データ(利用可能な場合)/当該施設で製造される全ての製品リスト
出所:INTEGRATION OF FDA FACILITY EVALUATION AND INSPECTION PROGRAM FOR HUMAN DRUGS: A CONCEPT OF OPERATION、じほう「世界をリードするFDAのGMP査察」、
FDA Field Alert Reports、FDA Biological Product Deviations
*1:査察官が査察を通じて観察した所見(指摘事項)を対象企業の経営陣に提示するもの、*2:出荷済医薬品の品質問題が発覚した際に医薬品の販売申請を行った者が
22
FDAに提出する報告書、*3 :生物学的製剤の製造に関する欠陥等が発覚した際に関連する製造所がFDAに提出する報告書