


































よむ、つかう、まなぶ。
【資料3】日米欧における医薬品の品質管理・製造管理に関する調査の状況(令和4年度欧米の薬事制度に関する調査・整理業務 調査結果) (30 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_30030.html |
| 出典情報 | 医薬品等行政評価・監視委員会(第10回 12/27)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
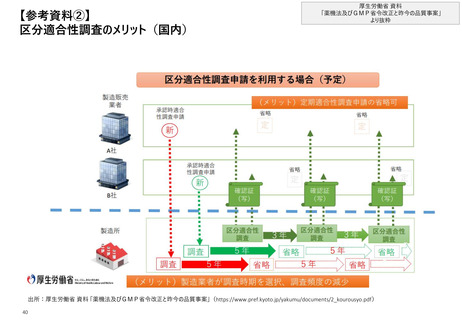
【4. 査察・調査の実施方法・内容】欧州では、リスクベース計画モデルに基づき定期的な
GMP査察が実施されており、査察対象に対して、リスクに応じて最大間隔を3年とした査察
頻度の設定、製品等に応じて査察にかかる所要日数が設定される
欧州_リスク評価
◼ 各規制当局は、年間の査察プログラムの準備、実現及び監督を網羅する手順を文書化し、計画どおりの範囲と頻度で査察を行う
◼ 医薬品製造業者に対する査察のためのリスクベース計画モデル(A model for risk based planning for inspections of pharmaceutical
manufacturers)は各規制当局が年間の査察プログラムを作成・実施するために整備されたモデルであり、GMP査察の頻度と範囲を計画する際に
活用できるシンプルな品質リスク管理ツールを提供することを目的としている
※ 定期的なGMP査察が対象。一度も査察を受けたことの無い製造業者に対する査察や、緊急の査察は対象外であり、そのような査察については公式なツールを
使う必要は原則無いと記載されている
◼ GMPの不遵守や重大な問題の発生を防ぐ観点から、通常、査察の間隔は3年を超えてはならないとされている
リスクベース計画モデルの運用方法
◼ 査察完了後すぐに、通常、査察官がワークシートを完成させる
※ワークシートにおいて、下記の項目の記載が必要
① 固有のリスクとコンプライアンス上のリスクを考慮したリスク評価
② ①の評価に基づく、推奨される査察頻度の設定
③ 次の定期的な査察の範囲の設定
◼ 所要時間の基準を参照し査察に必要な日数を設定する
※各規制当局のプログラム等に応じて日数は変更可とされているが、製造される
製品やプロセスに応じて以下のように基準が示されている
<所要時間の一例>
固有のリスクとコンプライアンス上のリスクを考慮したリスク評価
◼ 固有のリスクとコンプライアンス上のリスクを評価した上で、マトリック
スで最終的なリスク評価(リスクが低い順にA、B、C)を行う
施設やプロセス、製品の複雑さ及び施設が提供
する製品や業務の重要性に応じて評価
最近の不備状況
に応じて評価
コンプライアンス
上のリスク
低
中
高
固有のリスク
低
中
高
A
A
B
A
B
C
B
C
C
推奨される査察頻度の設定
◼ リスク評価を元に、下記のとおり査察頻度を設定する
• A:2年から3年に一度、B:1年から2年に一度、C:1年未満
製造している製品
査察全体の所要日数
無菌製剤
10日以上
非滅菌製剤
4日以上
次の定期的な査察の範囲の設定
生物学的製剤
7日以上
◼ 次の査察の焦点と深さ(査察において不備が確認された領域等を
考慮して決定)、必要な期間、査察官の人数、査察チームにおけ
る特定の能力や専門知識の要否を設定する
◼ 施設のコンプライアンスの遵守状況や活動、製品について新しい情
報を受け取った場合には、頻度や次の定期査察の範囲を更新する
出所:EMA Compilation of Union Procedures on Inspections and Exchange of Information (https://www.ema.europa.eu/en/documents/regulatory-procedural30 guideline/compilation-union-procedures-inspections-exchange-information_en.pdf)
GMP査察が実施されており、査察対象に対して、リスクに応じて最大間隔を3年とした査察
頻度の設定、製品等に応じて査察にかかる所要日数が設定される
欧州_リスク評価
◼ 各規制当局は、年間の査察プログラムの準備、実現及び監督を網羅する手順を文書化し、計画どおりの範囲と頻度で査察を行う
◼ 医薬品製造業者に対する査察のためのリスクベース計画モデル(A model for risk based planning for inspections of pharmaceutical
manufacturers)は各規制当局が年間の査察プログラムを作成・実施するために整備されたモデルであり、GMP査察の頻度と範囲を計画する際に
活用できるシンプルな品質リスク管理ツールを提供することを目的としている
※ 定期的なGMP査察が対象。一度も査察を受けたことの無い製造業者に対する査察や、緊急の査察は対象外であり、そのような査察については公式なツールを
使う必要は原則無いと記載されている
◼ GMPの不遵守や重大な問題の発生を防ぐ観点から、通常、査察の間隔は3年を超えてはならないとされている
リスクベース計画モデルの運用方法
◼ 査察完了後すぐに、通常、査察官がワークシートを完成させる
※ワークシートにおいて、下記の項目の記載が必要
① 固有のリスクとコンプライアンス上のリスクを考慮したリスク評価
② ①の評価に基づく、推奨される査察頻度の設定
③ 次の定期的な査察の範囲の設定
◼ 所要時間の基準を参照し査察に必要な日数を設定する
※各規制当局のプログラム等に応じて日数は変更可とされているが、製造される
製品やプロセスに応じて以下のように基準が示されている
<所要時間の一例>
固有のリスクとコンプライアンス上のリスクを考慮したリスク評価
◼ 固有のリスクとコンプライアンス上のリスクを評価した上で、マトリック
スで最終的なリスク評価(リスクが低い順にA、B、C)を行う
施設やプロセス、製品の複雑さ及び施設が提供
する製品や業務の重要性に応じて評価
最近の不備状況
に応じて評価
コンプライアンス
上のリスク
低
中
高
固有のリスク
低
中
高
A
A
B
A
B
C
B
C
C
推奨される査察頻度の設定
◼ リスク評価を元に、下記のとおり査察頻度を設定する
• A:2年から3年に一度、B:1年から2年に一度、C:1年未満
製造している製品
査察全体の所要日数
無菌製剤
10日以上
非滅菌製剤
4日以上
次の定期的な査察の範囲の設定
生物学的製剤
7日以上
◼ 次の査察の焦点と深さ(査察において不備が確認された領域等を
考慮して決定)、必要な期間、査察官の人数、査察チームにおけ
る特定の能力や専門知識の要否を設定する
◼ 施設のコンプライアンスの遵守状況や活動、製品について新しい情
報を受け取った場合には、頻度や次の定期査察の範囲を更新する
出所:EMA Compilation of Union Procedures on Inspections and Exchange of Information (https://www.ema.europa.eu/en/documents/regulatory-procedural30 guideline/compilation-union-procedures-inspections-exchange-information_en.pdf)