


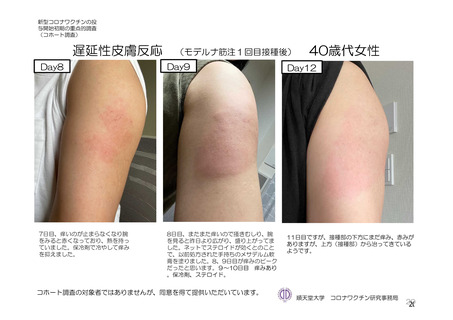
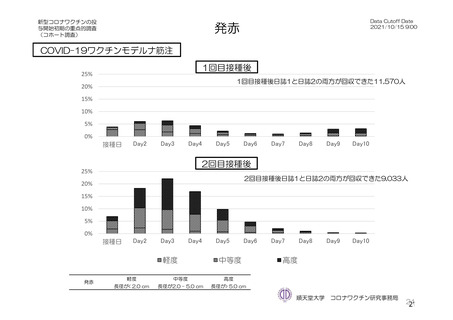
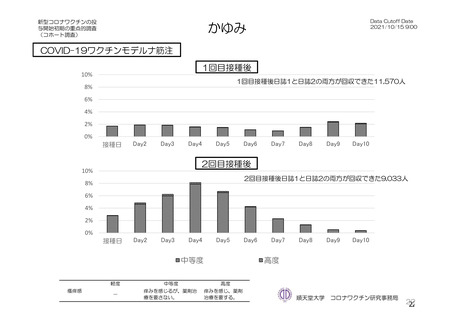
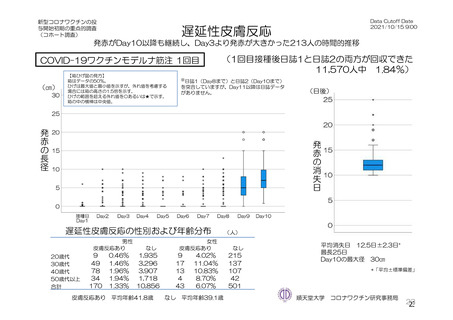





















































































































よむ、つかう、まなぶ。
参考資料1-7 浜口班の議論における参考資料(令和3年10月25日開催)(令和3年度第6回安全技術調査会参考資料1-2) (144 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_27504.html |
| 出典情報 | 薬事・食品衛生審議会 薬事分科会血液事業部会安全技術調査会(令和4年度第2回 8/23)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
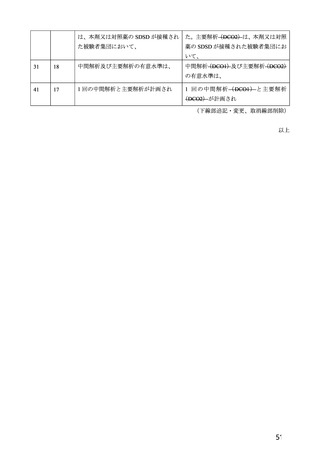
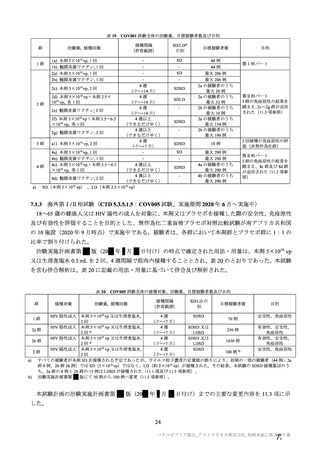
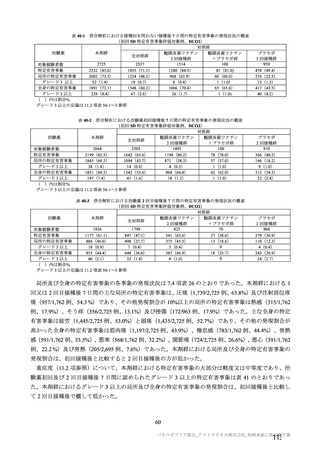
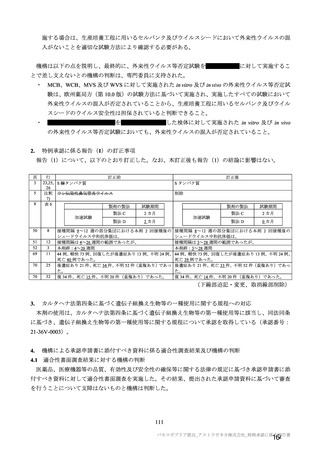
験で 18 歳以上及び COV005 試験で 18~65 歳であった。55~65 歳の成人及び 65 歳以上の高齢者の被験
者数は限られるものの、7.R.2.2.3 項、7.R.3.3.2 項及び 7.R.4.2.2 項における検討内容を踏まえると、本剤
の接種対象年齢は 18 歳以上とすることが適切と考える。
以上より、本剤の用法及び用量に関連する注意において、接種対象者として「本剤の接種は 18 歳以上
の者に行う。」と記載することが適切と考える。
7.R.7
製造販売後の検討事項について
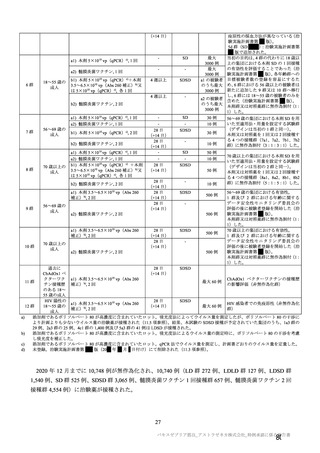
申請者は、本剤の製造販売後調査について、以下のように説明している。
本剤の長期データを含む日本人の安全性について、製造販売承認時までに得られる情報は限定的であ
ること等から(7.R.3 参照)、本剤の最終接種 12 カ月後までの安全性を検討することを目的とした一般
使用成績調査を実施する予定である。調査対象は、「新型コロナワクチンの投与開始初期の重点的調査
(コホート調査)」(令和 2 年度厚生労働行政推進調査事業費補助金
新興・再興感染症及び予防接種
政策推進研究事業、調査予定例数:臨時接種の対象となるワクチンについて各 10,000~20,000 例)に参
加する被接種者のうち、本剤の最終接種 12 カ月後までの追跡調査への参加に同意するすべての被接種
者とし、ショック・アナフィラキシー、免疫介在性の神経学的反応、及びワクチン関連の呼吸器疾患増
強(VAERD)を含むワクチン接種に伴う疾患増強(VAED)を安全性検討事項として設定する。観察期
間は、本剤の最終接種 28 日後(先行するコホート調査の観察期間終了日)の翌日から最終接種 12 カ月
後(11 カ月間)とする予定である。
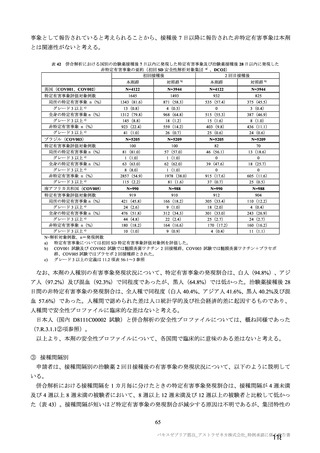
また、本剤の臨床試験で十分な安全性情報が得られていない、高齢者を含め COVID-19 の重症化リス
クが高いと考えられる基礎疾患を有する者(7.R.3.3.1 及び 7.R.4.2.1 参照)を対象に、本剤接種後の安全
性を検討する特定使用成績調査(観察期間:本剤初回接種日(1 日目)から最終接種後 28 日まで)を実
施する予定である。調査予定例数については、対象となる COVID-19 の重症化リスクが高いと考えられ
る者への接種時期が限定されることから、実施可能性の観点も踏まえて 1,000 例(安全性解析対象症例
として)と設定し、調査対象者のうち、重症化リスクが特に高いと考えられる高齢者(7.R.4.2.2 参照)
を一定例数組み入れる予定である。
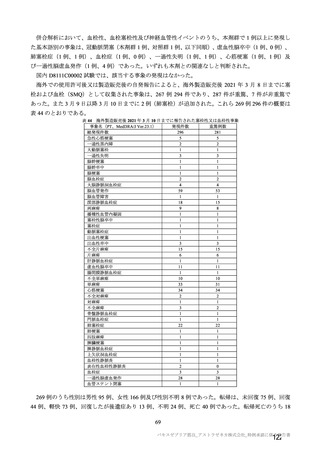
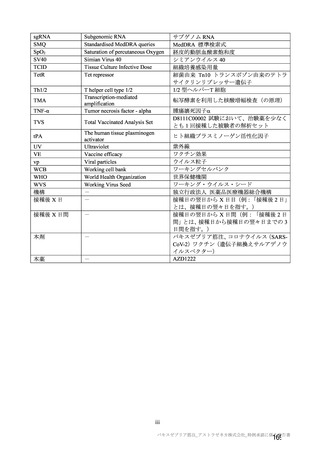
上記の調査における検討に加え、本剤承認後に国内 D8111C00002 試験から切り替えて実施する製造販
売後臨床試験、並びに COV001 試験、COV002 試験、COV003 試験、COV005 試験及び D8110C00001 試
験のフォローアップ等の情報も踏まえて本剤の長期の安全性等を検討する予定である。
また、本剤の適正使用を促し安全性の確保を図るため、追加のリスク最小化活動として、本剤の副反
応集計一覧を一定期間毎に作成し、医療従事者に提供する予定である。
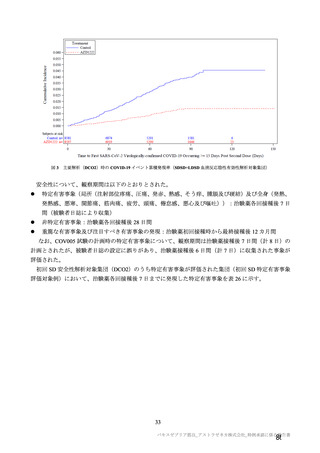
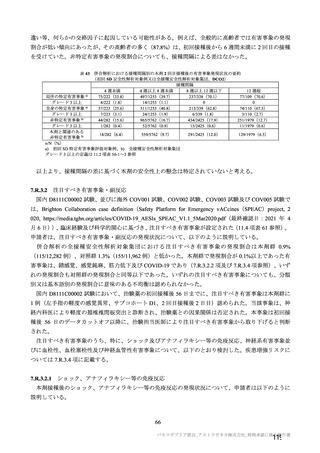
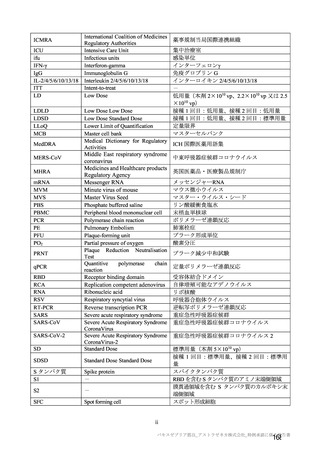
機構は、本剤接種後に血栓性、血栓塞栓性及び神経血管性イベントが認められていることから(7.R.3.3
参照)、当該事象についても安全性検討事項として設定し、製造販売後調査において発現状況を確認す
る必要があると考える。また、既承認の SARS-CoV-2 ワクチンの接種の進捗状況によって、本剤の接種
対象は変わりうるため、調査開始時点での本剤の接種対象を確認した上で、製造販売後調査計画の詳細
については適宜再検討する必要があると考える。
製造販売後の検討事項については、専門協議の議論も踏まえて最終的に判断したい。
カルタヘナ法第四条に基づく遺伝子組換え生物等の一種使用に関する規程への対応
現在、審査中であり、その結果及び機構の判断は報告(2)で報告する。
91
バキスゼブリア筋注_アストラゼネカ株式会社_特例承認に係る報告書
144
者数は限られるものの、7.R.2.2.3 項、7.R.3.3.2 項及び 7.R.4.2.2 項における検討内容を踏まえると、本剤
の接種対象年齢は 18 歳以上とすることが適切と考える。
以上より、本剤の用法及び用量に関連する注意において、接種対象者として「本剤の接種は 18 歳以上
の者に行う。」と記載することが適切と考える。
7.R.7
製造販売後の検討事項について
申請者は、本剤の製造販売後調査について、以下のように説明している。
本剤の長期データを含む日本人の安全性について、製造販売承認時までに得られる情報は限定的であ
ること等から(7.R.3 参照)、本剤の最終接種 12 カ月後までの安全性を検討することを目的とした一般
使用成績調査を実施する予定である。調査対象は、「新型コロナワクチンの投与開始初期の重点的調査
(コホート調査)」(令和 2 年度厚生労働行政推進調査事業費補助金
新興・再興感染症及び予防接種
政策推進研究事業、調査予定例数:臨時接種の対象となるワクチンについて各 10,000~20,000 例)に参
加する被接種者のうち、本剤の最終接種 12 カ月後までの追跡調査への参加に同意するすべての被接種
者とし、ショック・アナフィラキシー、免疫介在性の神経学的反応、及びワクチン関連の呼吸器疾患増
強(VAERD)を含むワクチン接種に伴う疾患増強(VAED)を安全性検討事項として設定する。観察期
間は、本剤の最終接種 28 日後(先行するコホート調査の観察期間終了日)の翌日から最終接種 12 カ月
後(11 カ月間)とする予定である。
また、本剤の臨床試験で十分な安全性情報が得られていない、高齢者を含め COVID-19 の重症化リス
クが高いと考えられる基礎疾患を有する者(7.R.3.3.1 及び 7.R.4.2.1 参照)を対象に、本剤接種後の安全
性を検討する特定使用成績調査(観察期間:本剤初回接種日(1 日目)から最終接種後 28 日まで)を実
施する予定である。調査予定例数については、対象となる COVID-19 の重症化リスクが高いと考えられ
る者への接種時期が限定されることから、実施可能性の観点も踏まえて 1,000 例(安全性解析対象症例
として)と設定し、調査対象者のうち、重症化リスクが特に高いと考えられる高齢者(7.R.4.2.2 参照)
を一定例数組み入れる予定である。
上記の調査における検討に加え、本剤承認後に国内 D8111C00002 試験から切り替えて実施する製造販
売後臨床試験、並びに COV001 試験、COV002 試験、COV003 試験、COV005 試験及び D8110C00001 試
験のフォローアップ等の情報も踏まえて本剤の長期の安全性等を検討する予定である。
また、本剤の適正使用を促し安全性の確保を図るため、追加のリスク最小化活動として、本剤の副反
応集計一覧を一定期間毎に作成し、医療従事者に提供する予定である。
機構は、本剤接種後に血栓性、血栓塞栓性及び神経血管性イベントが認められていることから(7.R.3.3
参照)、当該事象についても安全性検討事項として設定し、製造販売後調査において発現状況を確認す
る必要があると考える。また、既承認の SARS-CoV-2 ワクチンの接種の進捗状況によって、本剤の接種
対象は変わりうるため、調査開始時点での本剤の接種対象を確認した上で、製造販売後調査計画の詳細
については適宜再検討する必要があると考える。
製造販売後の検討事項については、専門協議の議論も踏まえて最終的に判断したい。
カルタヘナ法第四条に基づく遺伝子組換え生物等の一種使用に関する規程への対応
現在、審査中であり、その結果及び機構の判断は報告(2)で報告する。
91
バキスゼブリア筋注_アストラゼネカ株式会社_特例承認に係る報告書
144