












































































よむ、つかう、まなぶ。
資料No.1-1~1-5_第十八改正日本薬局方第二追補(案) (34 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/shingi2/0000174942_00008.html |
| 出典情報 | 薬事・食品衛生審議会 日本薬局方部会(令和5年度第1回 1/22)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
6 カルメロースカルシウム
第十八改正日本薬局方第二追補
1
不溶性微粒子〈6.07〉
2
無菌 〈4.06〉
メンブランフィルター法により試験を行うとき, 47
3
適合する.
48
4
定量法
試験を行うとき,適合する.
本品のオキサリプラチン(C8H14N2O4Pt)約10 mgに対
5
応する容量を正確に量り,水を加えて正確に100 mLとし,
6
試料溶液とする.別にオキサリプラチン標準品(別途「オキ
46
49
◆
「
」で囲むことにより示す.
◆
三薬局方の調和合意に関する情報については,独立行政法人医薬
品医療機器総合機構のウェブサイトに掲載している.
確認試験
50
(4)
51
酸(31) 6 mLを加えて溶かし,必要ならばろ過し,煮沸した
52
後,冷却し,アンモニア試液で中和するとき,液はカルシウ
53
ム塩の定性反応〈1.09〉の(3)を呈する.
7
サリプラチン」と同様の方法で乾燥減量〈2.41〉を測定して
8
おく)約20 mgを精密に量り,水に溶かし正確に200 mLとし,
9
標準溶液とする.試料溶液及び標準溶液20 μLずつを正確に
10
とり,次の条件で液体クロマトグラフィー〈2.01〉 により試
54
純度試験
本品1 gを強熱して灰化し,残留物に水10 mL及び酢
11
験を行い,それぞれの液のオキサリプラチンのピーク面積
55
(3)
12
AT及びASを測定する.
56
場合に適用する.(2)の試料溶液10 mLに塩酸1 mLを加え,
13
オキサリプラチン(C8H14N2O4Pt)の量(mg)
57
水浴中で綿状の沈殿が生じるまで加熱し,冷却した後,遠心
58
分離する.上澄液をとり,沈殿を水10 mLずつで3回洗い,
14
=MS × AT/AS × 1/2
15
Ms:乾燥物に換算したオキサリプラチン標準品の秤取量
16
17
(mg)
試験条件
18
19
「オキサリプラチン」の定量法の試験条件を準用する.
システム適合性
20
システムの性能:オキサリプラチン溶液(1→500) 1 mL
毎回遠心分離し,上澄液及び洗液を合わせ,水を加えて100
60
mLとする.この液25 mLをとり,3 mol/L塩酸試液1 mL及
61
び水を加えて50 mLとし,検液とする.別に水25 mLに
62
0.005 mol/L硫酸0.42 mLを加え,更に3 mol/L塩酸試液1
63
mL及び水を加えて50 mLとし,比較液として試験を行う.
64
ただし,検液及び比較液には塩化バリウム試液3 mLずつを
65
加える(1.0%以下).
66
21
及び1 mol/L塩化ナトリウム試液1 mLを量り,水を加
えて10 mLとする.この液を60℃で約2時間加熱後,
23
放冷する.この液20 μLにつき,上記の条件で操作す
24
るとき,オキサリプラチンに対する相対保持時間約
67
25
0.9のピークとオキサリプラチンの分離度は2.0以上で
68
26
あり,オキサリプラチンのシンメトリー係数は2.0以
27
下である.
28
システムの再現性:標準溶液20 μLにつき,上記の条件
で試験を6回繰り返すとき,オキサリプラチンのピー
30
ク面積の相対標準偏差は1.0%以下である.
31
貯法 容器 密封容器.
32
その他
製造工程において硫酸が使用される
59
22
29
硫酸塩〈1.14〉
69
70
強熱残分〈2.44〉 10.0 ~ 20.0%(乾燥後,1 g).
医薬品各条の部
グリセリンの条純度試験の項ヒ素の目を削
り,以降を繰り上げる.
医薬品各条の部
濃グリセリンの条純度試験の項ヒ素の目を
削り,以降を繰り上げる.
33
類縁物質Bは,「オキサリプラチン」のその他を準用する.
71
34
類縁物質IA:
72
の項及び確認試験の項を次のように改める.
35
(SP-4-2)-Di-μ-oxobis[(1R,2R)-cyclohexane-1,2-diamine-
36
κN,κN']diplatinum
73
クリンダマイシンリン酸エステル
74
性状 本品は白色~微黄白色の結晶性の粉末である.
75
76
37
77
78
38
医薬品各条の部
カルメロースカルシウムの条冒頭の国際調
医薬品各条の部
クリンダマイシンリン酸エステルの条性状
本品は水に溶けやすく,メタノールにやや溶けにくく,エ
タノール(95)にほとんど溶けない.
本品は結晶多形が認められる.
確認試験
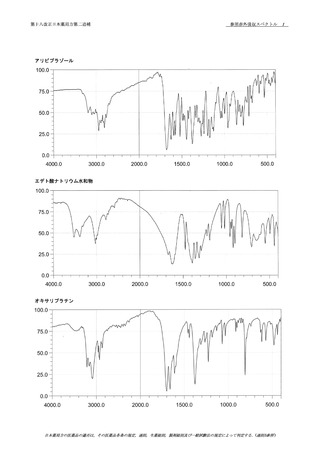
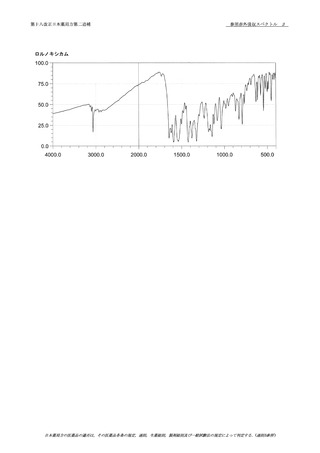
本品を100℃で2時間乾燥し,赤外吸収スペクトル
79
測定法〈2.25〉のペースト法又はATR法により試験を行い,
80
本品のスペクトルと100℃で2時間乾燥したクリンダマイシ
81
ンリン酸エステル標準品のスペクトルを比較するとき,両者
のスペクトルは同一波数のところに同様の強度の吸収を認め
39
和に関する記載,確認試験の項(4)の目,純度試験の項
40
(3)の目及び強熱残分の項を次のように改める.
82
83
る.もし,これらのスペクトルに差を認めるときは,本品及
41
カルメロースカルシウム
84
びクリンダマイシンリン酸エステル標準品50 mgずつをとり,
85
それぞれに水0.2 mLを加えて加熱して溶かし,蒸発乾固し
86
た後,残留物を100 ~ 105℃で2時間乾燥したものにつき,
87
同様の試験を行う.
42
43
本医薬品各条は,三薬局方での調和合意に基づき規定した医薬品
各条である.
44
なお,三薬局方で調和されていない部分のうち,調和合意におい
45
て,調和の対象とされた項中非調和となっている項の該当箇所は
日本薬局方の医薬品の適否は,その医薬品各条の規定,通則,生薬総則,製剤総則及び一般試験法の規定によって判定する.(通則5参照 )
第十八改正日本薬局方第二追補
1
不溶性微粒子〈6.07〉
2
無菌 〈4.06〉
メンブランフィルター法により試験を行うとき, 47
3
適合する.
48
4
定量法
試験を行うとき,適合する.
本品のオキサリプラチン(C8H14N2O4Pt)約10 mgに対
5
応する容量を正確に量り,水を加えて正確に100 mLとし,
6
試料溶液とする.別にオキサリプラチン標準品(別途「オキ
46
49
◆
「
」で囲むことにより示す.
◆
三薬局方の調和合意に関する情報については,独立行政法人医薬
品医療機器総合機構のウェブサイトに掲載している.
確認試験
50
(4)
51
酸(31) 6 mLを加えて溶かし,必要ならばろ過し,煮沸した
52
後,冷却し,アンモニア試液で中和するとき,液はカルシウ
53
ム塩の定性反応〈1.09〉の(3)を呈する.
7
サリプラチン」と同様の方法で乾燥減量〈2.41〉を測定して
8
おく)約20 mgを精密に量り,水に溶かし正確に200 mLとし,
9
標準溶液とする.試料溶液及び標準溶液20 μLずつを正確に
10
とり,次の条件で液体クロマトグラフィー〈2.01〉 により試
54
純度試験
本品1 gを強熱して灰化し,残留物に水10 mL及び酢
11
験を行い,それぞれの液のオキサリプラチンのピーク面積
55
(3)
12
AT及びASを測定する.
56
場合に適用する.(2)の試料溶液10 mLに塩酸1 mLを加え,
13
オキサリプラチン(C8H14N2O4Pt)の量(mg)
57
水浴中で綿状の沈殿が生じるまで加熱し,冷却した後,遠心
58
分離する.上澄液をとり,沈殿を水10 mLずつで3回洗い,
14
=MS × AT/AS × 1/2
15
Ms:乾燥物に換算したオキサリプラチン標準品の秤取量
16
17
(mg)
試験条件
18
19
「オキサリプラチン」の定量法の試験条件を準用する.
システム適合性
20
システムの性能:オキサリプラチン溶液(1→500) 1 mL
毎回遠心分離し,上澄液及び洗液を合わせ,水を加えて100
60
mLとする.この液25 mLをとり,3 mol/L塩酸試液1 mL及
61
び水を加えて50 mLとし,検液とする.別に水25 mLに
62
0.005 mol/L硫酸0.42 mLを加え,更に3 mol/L塩酸試液1
63
mL及び水を加えて50 mLとし,比較液として試験を行う.
64
ただし,検液及び比較液には塩化バリウム試液3 mLずつを
65
加える(1.0%以下).
66
21
及び1 mol/L塩化ナトリウム試液1 mLを量り,水を加
えて10 mLとする.この液を60℃で約2時間加熱後,
23
放冷する.この液20 μLにつき,上記の条件で操作す
24
るとき,オキサリプラチンに対する相対保持時間約
67
25
0.9のピークとオキサリプラチンの分離度は2.0以上で
68
26
あり,オキサリプラチンのシンメトリー係数は2.0以
27
下である.
28
システムの再現性:標準溶液20 μLにつき,上記の条件
で試験を6回繰り返すとき,オキサリプラチンのピー
30
ク面積の相対標準偏差は1.0%以下である.
31
貯法 容器 密封容器.
32
その他
製造工程において硫酸が使用される
59
22
29
硫酸塩〈1.14〉
69
70
強熱残分〈2.44〉 10.0 ~ 20.0%(乾燥後,1 g).
医薬品各条の部
グリセリンの条純度試験の項ヒ素の目を削
り,以降を繰り上げる.
医薬品各条の部
濃グリセリンの条純度試験の項ヒ素の目を
削り,以降を繰り上げる.
33
類縁物質Bは,「オキサリプラチン」のその他を準用する.
71
34
類縁物質IA:
72
の項及び確認試験の項を次のように改める.
35
(SP-4-2)-Di-μ-oxobis[(1R,2R)-cyclohexane-1,2-diamine-
36
κN,κN']diplatinum
73
クリンダマイシンリン酸エステル
74
性状 本品は白色~微黄白色の結晶性の粉末である.
75
76
37
77
78
38
医薬品各条の部
カルメロースカルシウムの条冒頭の国際調
医薬品各条の部
クリンダマイシンリン酸エステルの条性状
本品は水に溶けやすく,メタノールにやや溶けにくく,エ
タノール(95)にほとんど溶けない.
本品は結晶多形が認められる.
確認試験
本品を100℃で2時間乾燥し,赤外吸収スペクトル
79
測定法〈2.25〉のペースト法又はATR法により試験を行い,
80
本品のスペクトルと100℃で2時間乾燥したクリンダマイシ
81
ンリン酸エステル標準品のスペクトルを比較するとき,両者
のスペクトルは同一波数のところに同様の強度の吸収を認め
39
和に関する記載,確認試験の項(4)の目,純度試験の項
40
(3)の目及び強熱残分の項を次のように改める.
82
83
る.もし,これらのスペクトルに差を認めるときは,本品及
41
カルメロースカルシウム
84
びクリンダマイシンリン酸エステル標準品50 mgずつをとり,
85
それぞれに水0.2 mLを加えて加熱して溶かし,蒸発乾固し
86
た後,残留物を100 ~ 105℃で2時間乾燥したものにつき,
87
同様の試験を行う.
42
43
本医薬品各条は,三薬局方での調和合意に基づき規定した医薬品
各条である.
44
なお,三薬局方で調和されていない部分のうち,調和合意におい
45
て,調和の対象とされた項中非調和となっている項の該当箇所は
日本薬局方の医薬品の適否は,その医薬品各条の規定,通則,生薬総則,製剤総則及び一般試験法の規定によって判定する.(通則5参照 )