










































































































よむ、つかう、まなぶ。
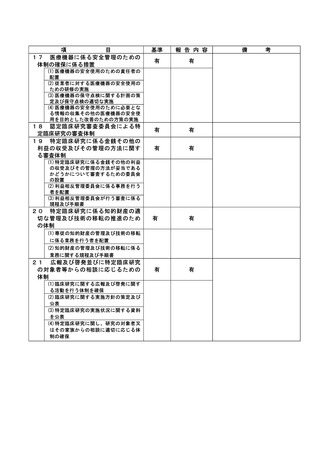
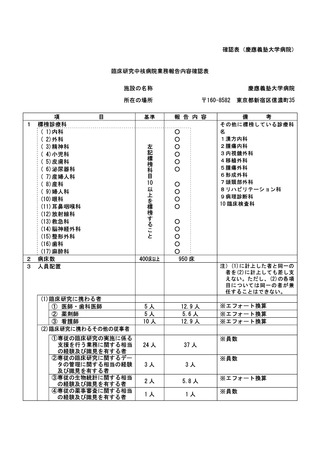
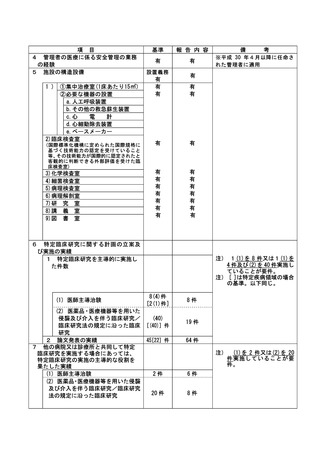
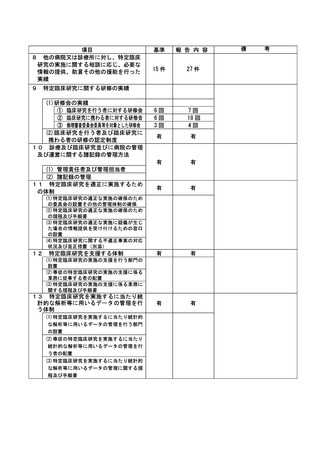
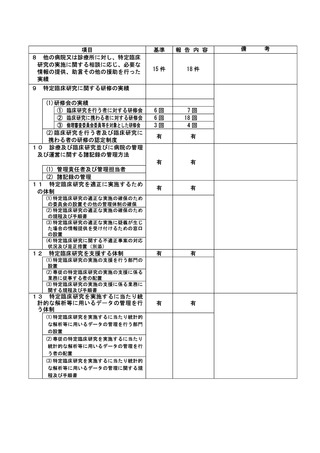
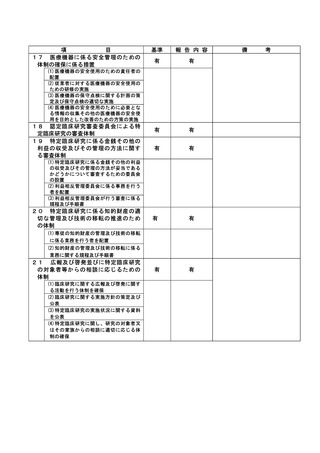
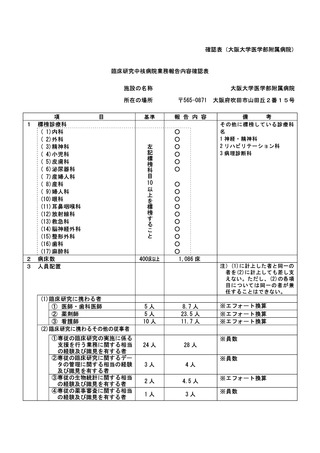
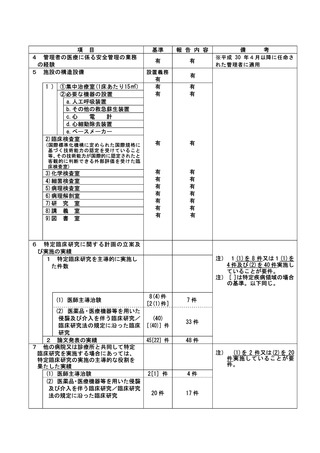
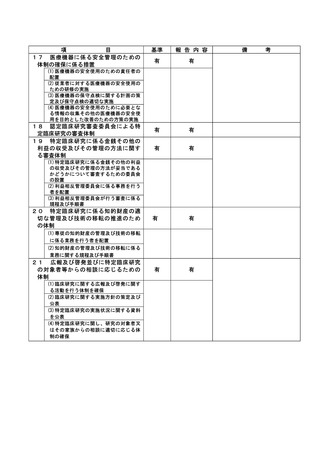
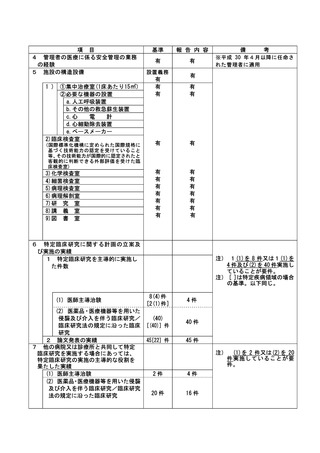
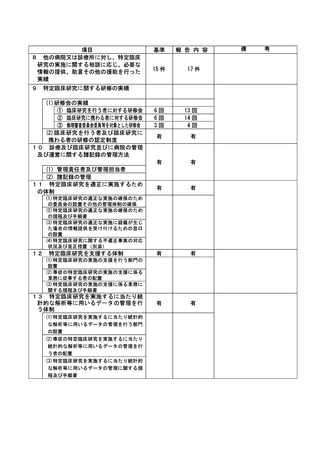
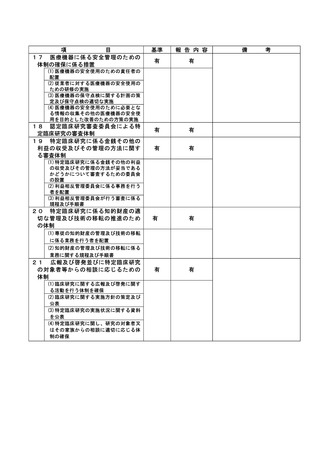
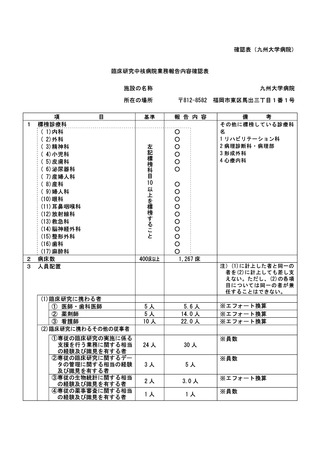
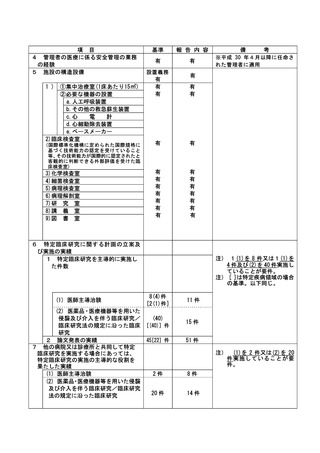
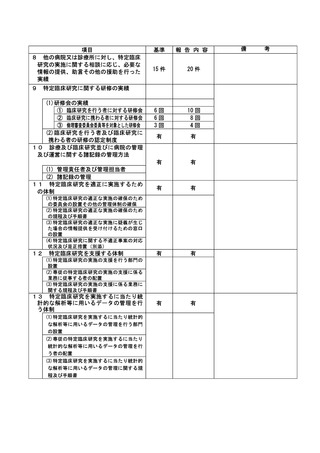
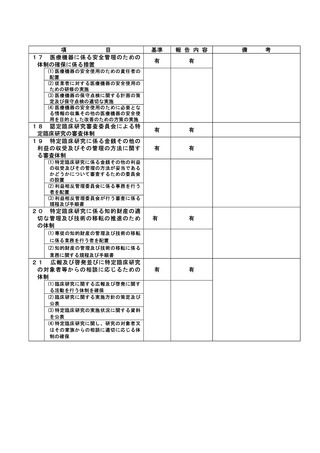
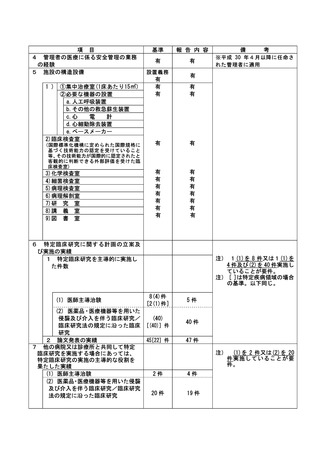
資料2:臨床研究中核病院業務報告内容確認表 (132 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_42147.html |
| 出典情報 | 厚生科学審議会 臨床研究部会(第35回 8/8)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
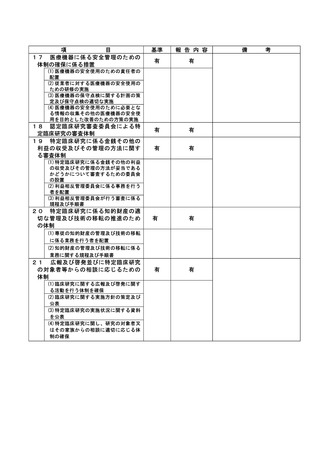
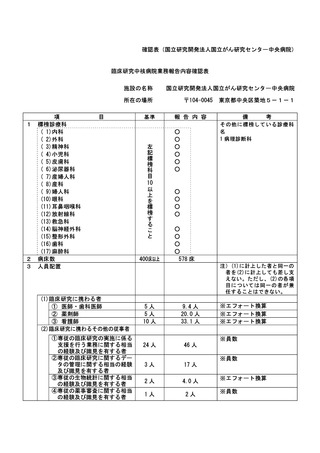
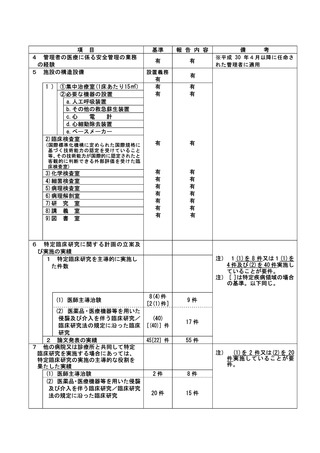
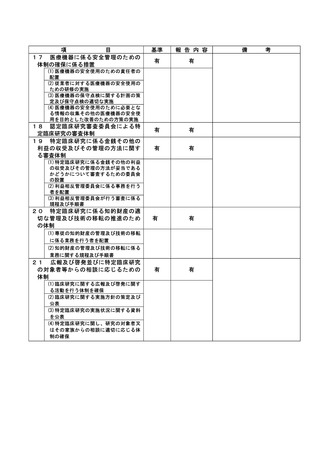
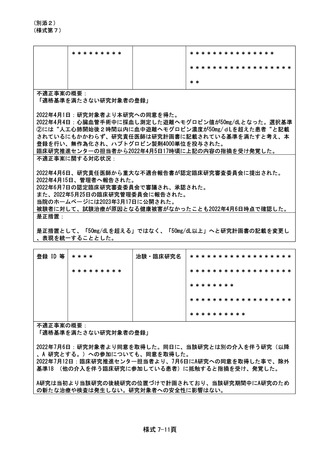
(様式第7)
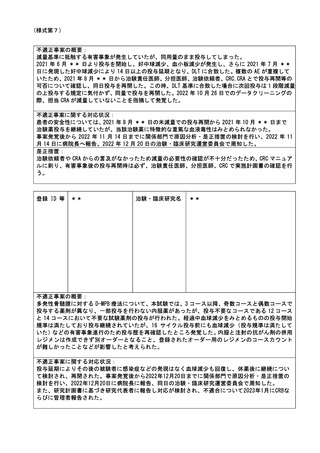
検査項目は、研究計画書上は観察項目の一項目であったが主要評価項目ではなく、またスピッツは異なっ
ていたが項目の測定は可能であったため、追加の採血は行っていない。
是正措置:
企業から提供の資材の内容に間違いがあったことに気づかなかったことが原因であったが、研究協力者は
事務職員であり、スピッツの間違いを事前に発見することができなかった。事案を*****社に報告
し、本研究に限らず提供資材の確認徹底(再発防止)を要請した。
また、提供資材に間違いがないよう、複数人で確認を徹底する体制とした。
登録 ID 等
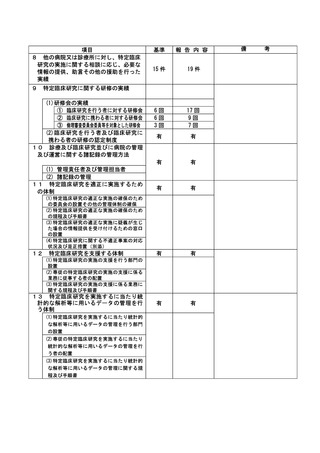
*******
治験・臨床研究名
******************
******************
不適正事案の概要:
発生日:2023/2/7
患者は治験の同意後に前治療*****の最終投与(登録日の 27 日前)を受けていた。スクリーニング
検査翌日に治験の登録を行い、登録から 5 日目に治験製品の投与を開始した。
本治験では、プロトコールの除外基準に「登録前 4 週間以内に抗腫瘍薬の投与を受けた患者」が定義され
ており、モニタリングの際に担当モニターより*****投与から登録までの期間が 4 週間以内(27 日)
になっていることを指摘され逸脱が発覚した。
不適正事案に関する対応状況:
責任医師は、臨床上問題はなく治験の継続にも影響がないと判断した。また、逸脱の経緯について、電子
カルテ内に治験責任医師が記載した。
是正措置:
適格性確認時に医師、CRCともに*****の最終投与日から治験製品投与まで4週間空いていれば問題が
ないと思い込んでいたことが原因として挙げられる。
再発防止策として、医師、CRC、担当モニター間で事前にスケジュールを共有し、登録予定日や投与開始
日を調整する。
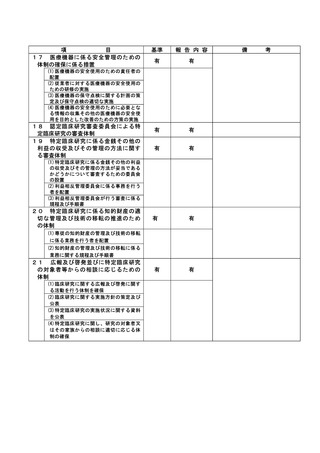
登録 ID 等
*******
治験・臨床研究名
******************
******************
不適正事案の概要:
発生日:2022/9/27
患者は後観察期間に移行、28日毎に規定visit通院中であり、原疾患の悪化に対して他施設で追加治療を
検討していた。規定visitから1週間後、他施設からの連絡により分担医師は患者の入院予定を把握しカル
テに記載していたが、CRCへの報告は無かった。後日CRCがカルテを確認していた際に発覚。既に入院日か
ら15日が経過していた。
プロトコール規定では後観察期間終了までが SAE 報告の対象期間であった。原疾患の悪化による追加治療
のための入院も SAE の報告対象であり、逸脱となった。
不適正事案に関する対応状況:
原疾患の悪化に対する後治療目的の入院であり、治験製品との因果関係は無いことを確認した。
CRC が知り得た翌日に SAE 第 1 報を提出した。逸脱の詳細については、CRC が報告書案を作成し、治験分
担医師が確認して保管した。
是正措置:
分担医師に他施設への入院がSAEに該当するという認識がなかったこと、後治療で入院した場合に当院に
報告する旨の患者指導や医師間の連携が不足していたことが原因として挙げられる。
再発防止策して、他施設での治療が検討された時点で、入院がSAE報告の対象となり得ることを分担医師
と共有する。患者家族や他院医師にも報告の必要性について再度説明する。
【様式第7】11
131
検査項目は、研究計画書上は観察項目の一項目であったが主要評価項目ではなく、またスピッツは異なっ
ていたが項目の測定は可能であったため、追加の採血は行っていない。
是正措置:
企業から提供の資材の内容に間違いがあったことに気づかなかったことが原因であったが、研究協力者は
事務職員であり、スピッツの間違いを事前に発見することができなかった。事案を*****社に報告
し、本研究に限らず提供資材の確認徹底(再発防止)を要請した。
また、提供資材に間違いがないよう、複数人で確認を徹底する体制とした。
登録 ID 等
*******
治験・臨床研究名
******************
******************
不適正事案の概要:
発生日:2023/2/7
患者は治験の同意後に前治療*****の最終投与(登録日の 27 日前)を受けていた。スクリーニング
検査翌日に治験の登録を行い、登録から 5 日目に治験製品の投与を開始した。
本治験では、プロトコールの除外基準に「登録前 4 週間以内に抗腫瘍薬の投与を受けた患者」が定義され
ており、モニタリングの際に担当モニターより*****投与から登録までの期間が 4 週間以内(27 日)
になっていることを指摘され逸脱が発覚した。
不適正事案に関する対応状況:
責任医師は、臨床上問題はなく治験の継続にも影響がないと判断した。また、逸脱の経緯について、電子
カルテ内に治験責任医師が記載した。
是正措置:
適格性確認時に医師、CRCともに*****の最終投与日から治験製品投与まで4週間空いていれば問題が
ないと思い込んでいたことが原因として挙げられる。
再発防止策として、医師、CRC、担当モニター間で事前にスケジュールを共有し、登録予定日や投与開始
日を調整する。
登録 ID 等
*******
治験・臨床研究名
******************
******************
不適正事案の概要:
発生日:2022/9/27
患者は後観察期間に移行、28日毎に規定visit通院中であり、原疾患の悪化に対して他施設で追加治療を
検討していた。規定visitから1週間後、他施設からの連絡により分担医師は患者の入院予定を把握しカル
テに記載していたが、CRCへの報告は無かった。後日CRCがカルテを確認していた際に発覚。既に入院日か
ら15日が経過していた。
プロトコール規定では後観察期間終了までが SAE 報告の対象期間であった。原疾患の悪化による追加治療
のための入院も SAE の報告対象であり、逸脱となった。
不適正事案に関する対応状況:
原疾患の悪化に対する後治療目的の入院であり、治験製品との因果関係は無いことを確認した。
CRC が知り得た翌日に SAE 第 1 報を提出した。逸脱の詳細については、CRC が報告書案を作成し、治験分
担医師が確認して保管した。
是正措置:
分担医師に他施設への入院がSAEに該当するという認識がなかったこと、後治療で入院した場合に当院に
報告する旨の患者指導や医師間の連携が不足していたことが原因として挙げられる。
再発防止策して、他施設での治療が検討された時点で、入院がSAE報告の対象となり得ることを分担医師
と共有する。患者家族や他院医師にも報告の必要性について再度説明する。
【様式第7】11
131