



































































































よむ、つかう、まなぶ。
別紙1○【先進医療会議】先進医療Bに係る新規技術の科学的評価等について (77 ページ)
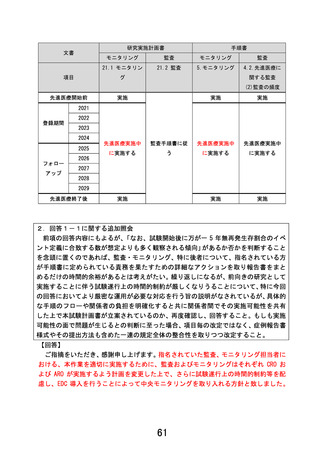
出典
| 公開元URL | https://www.mhlw.go.jp/stf/shingi2/0000205617_00053.html |
| 出典情報 | 先進医療会議(第117回先進医療会議、第142回先進医療技術審査部会 12/8)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
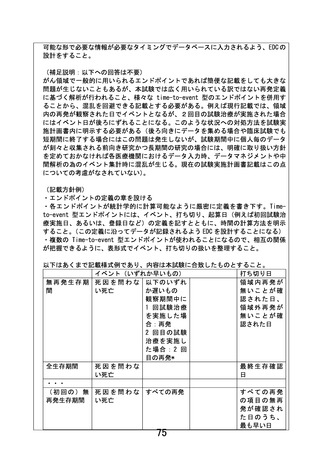
本試験は、後ろ向きの観察研究ではなく前向きの研究で有効性・安全性の確立して
いない医療行為を多施設共同試験の形で長期にわたり検討するものであることか
ら、有害事象の発現状況や試験治療の実施状況、場合によっては逸脱の発生状況と
その内容・対処方法を適時全施設で共有する必要がある。例えば Grade1 や 2 の直腸
瘻(尿道直腸瘻)が発生した場合、それ自体は重篤な有害事象として扱われない
が、それが散発するようであれば本技術のリスク・ベネフィット評価に影響を与え
うる。また、登録される被験者に対して安全に試験治療を行うためにも、適格基
準、術者規定、試験治療実施基準、試験治療実施方法等に見直すべき点はないか等
を考察し、施設間で共有することが被験者に対するリスクを小さくするために必要
となる。そのような検討を行うという観点での試験期間中の管理方法が不明瞭であ
るので追記が必要との趣旨である。
【回答】
(1)“21.品質管理及び品質保証に関する事項”にてデータマネジメントおよびモニ
タリングに関する記載を致しました。
また、中央モニタリングに関しましては、データマネジメント手順書に「中央モ
ニタリング等で安全性・逸脱等を確認した場合、研究責任医師に報告する」旨の
記載を致します。
また効果安全性評価委員会に関連する流れは、“8.3.5.中間解析結果の報告と審
査”に下記のように記載いたしました。
“中間解析の結果、効果・安全性評価委員会より試験の中止または変更の勧告が
なされた場合、研究事務局/研究代表医師/研究責任医師/統計担当共同研究者/研
究・開発計画支援担当研究者の合意のもとで試験の中止または変更を行うかどう
かを決定する。効果・安全性評価委員会より試験の中止または変更の勧告がなさ
れ、プロトコール内容の変更を行って試験を継続する場合は、プロトコール改
訂・改正が承認されるまでは全施設からの登録を一時中止する。”
(2) ご指摘に従い、「27.本研究の中止基準」表過去に報告された有害事象とその頻度
の末尾に、“すなわち、尿道直腸瘻については、当初の予測を大幅に上回るか否かの
判断は 2.6%ではなく 0.93%となる”ことを追記いたしました。
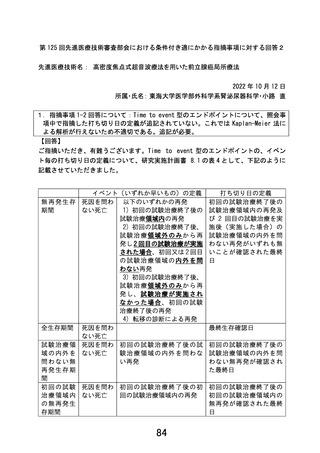
4.事前照会回答7-4に関して
(1)EDC へのデータ入力について、本試験において、参加医療機関側でどのようなタ
イミングでどのような入力作業が必要かを試験実施計画書内に明示すること。スケ
ジュール表はあくまで簡便な情報であり、各タイミングで提出すべき帳票の種類を
本文中で定義すること。例えば primary endpoint に関わる再発の情報は 48 週以降
どのようなタイミングで評価されるかが定義されていない。
(2)また、手順書等の試験実施計画書以外の文書の整備には一定の意義があるが、試
験実施計画書には参加医療機関側が取るべきアクションが全て記載されている必要
がある。手順書・マニュアル等は各々のアクションの詳細を解説するものと位置づ
けること。
(補足説明:以下への回答は不要)
77
いない医療行為を多施設共同試験の形で長期にわたり検討するものであることか
ら、有害事象の発現状況や試験治療の実施状況、場合によっては逸脱の発生状況と
その内容・対処方法を適時全施設で共有する必要がある。例えば Grade1 や 2 の直腸
瘻(尿道直腸瘻)が発生した場合、それ自体は重篤な有害事象として扱われない
が、それが散発するようであれば本技術のリスク・ベネフィット評価に影響を与え
うる。また、登録される被験者に対して安全に試験治療を行うためにも、適格基
準、術者規定、試験治療実施基準、試験治療実施方法等に見直すべき点はないか等
を考察し、施設間で共有することが被験者に対するリスクを小さくするために必要
となる。そのような検討を行うという観点での試験期間中の管理方法が不明瞭であ
るので追記が必要との趣旨である。
【回答】
(1)“21.品質管理及び品質保証に関する事項”にてデータマネジメントおよびモニ
タリングに関する記載を致しました。
また、中央モニタリングに関しましては、データマネジメント手順書に「中央モ
ニタリング等で安全性・逸脱等を確認した場合、研究責任医師に報告する」旨の
記載を致します。
また効果安全性評価委員会に関連する流れは、“8.3.5.中間解析結果の報告と審
査”に下記のように記載いたしました。
“中間解析の結果、効果・安全性評価委員会より試験の中止または変更の勧告が
なされた場合、研究事務局/研究代表医師/研究責任医師/統計担当共同研究者/研
究・開発計画支援担当研究者の合意のもとで試験の中止または変更を行うかどう
かを決定する。効果・安全性評価委員会より試験の中止または変更の勧告がなさ
れ、プロトコール内容の変更を行って試験を継続する場合は、プロトコール改
訂・改正が承認されるまでは全施設からの登録を一時中止する。”
(2) ご指摘に従い、「27.本研究の中止基準」表過去に報告された有害事象とその頻度
の末尾に、“すなわち、尿道直腸瘻については、当初の予測を大幅に上回るか否かの
判断は 2.6%ではなく 0.93%となる”ことを追記いたしました。
4.事前照会回答7-4に関して
(1)EDC へのデータ入力について、本試験において、参加医療機関側でどのようなタ
イミングでどのような入力作業が必要かを試験実施計画書内に明示すること。スケ
ジュール表はあくまで簡便な情報であり、各タイミングで提出すべき帳票の種類を
本文中で定義すること。例えば primary endpoint に関わる再発の情報は 48 週以降
どのようなタイミングで評価されるかが定義されていない。
(2)また、手順書等の試験実施計画書以外の文書の整備には一定の意義があるが、試
験実施計画書には参加医療機関側が取るべきアクションが全て記載されている必要
がある。手順書・マニュアル等は各々のアクションの詳細を解説するものと位置づ
けること。
(補足説明:以下への回答は不要)
77