














































































































































よむ、つかう、まなぶ。
【資料No.1】2.5_臨床に関する概括資料 (196 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_29325.html |
| 出典情報 | 薬事・食品衛生審議会 薬事分科会(令和4年度第5回 11/22)、医薬品第二部会(令和4年度第13回 11/22)(合同開催)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
S-217622
2.5.5.2.8
2.5 臨床に関する概括評価
安全性評価のまとめ
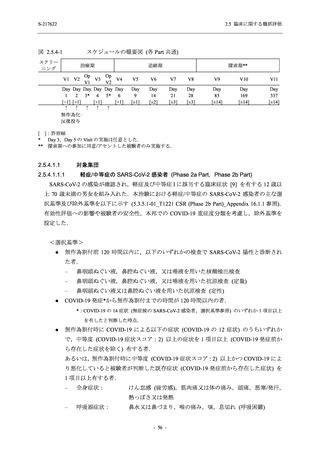
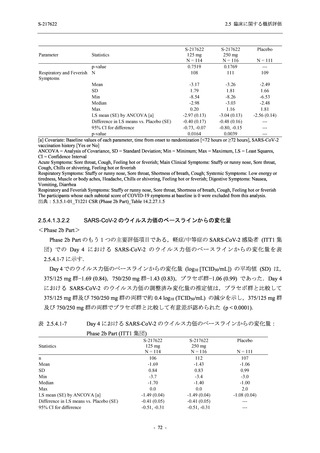
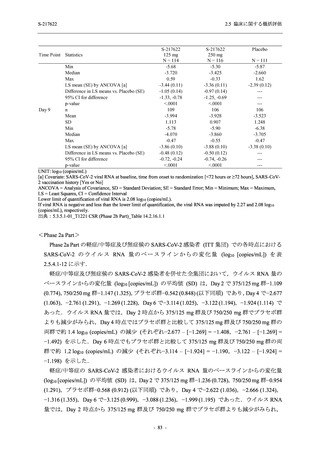
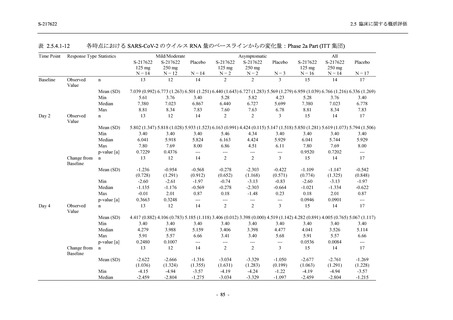
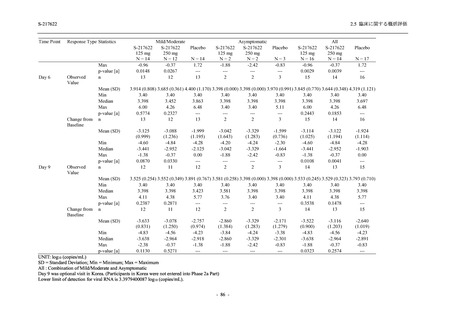
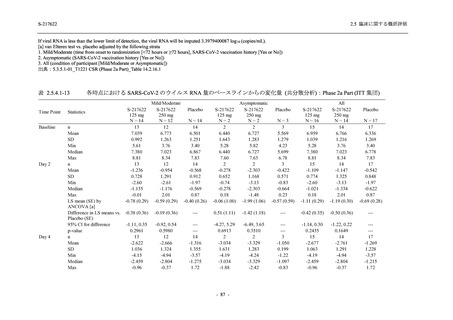
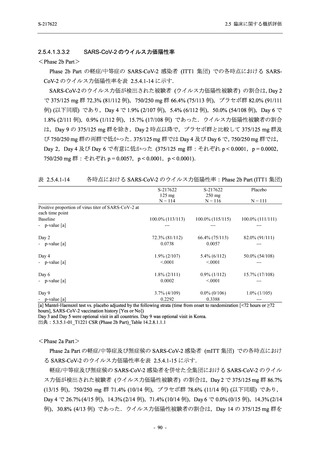
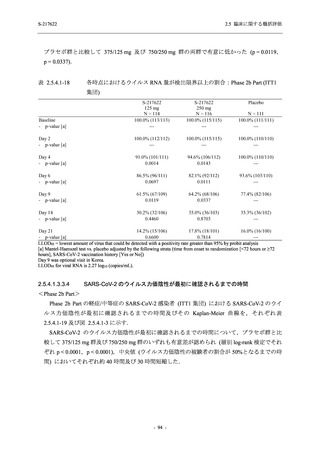
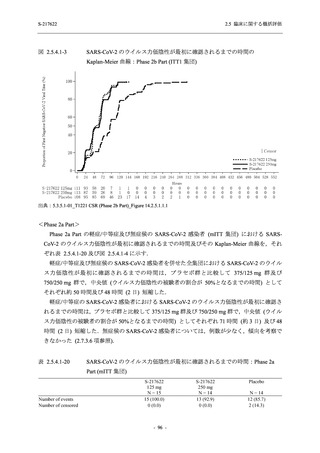
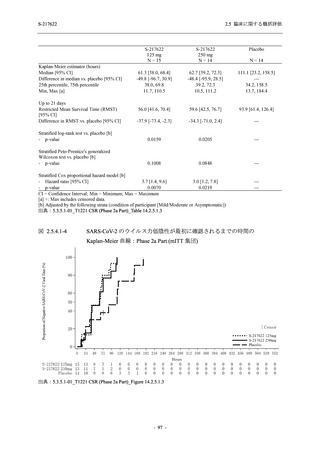
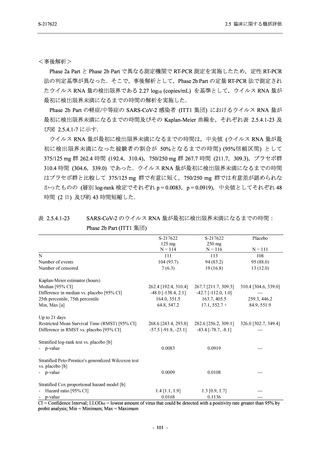
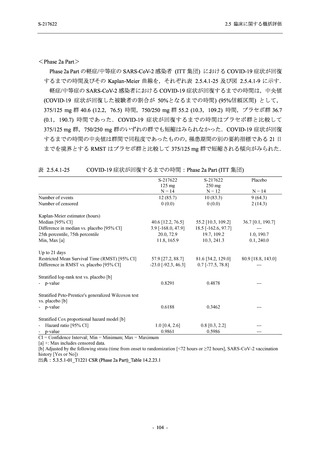
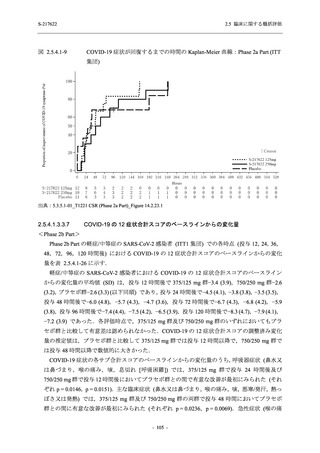
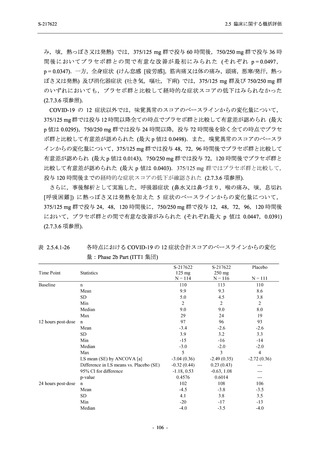
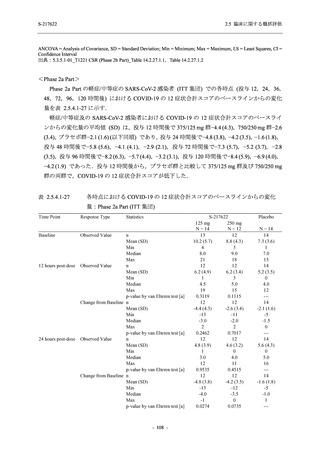
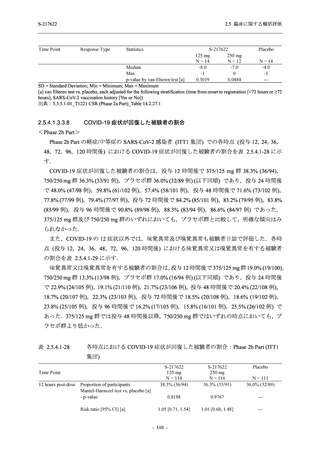
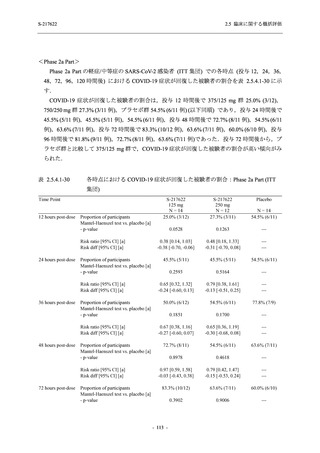
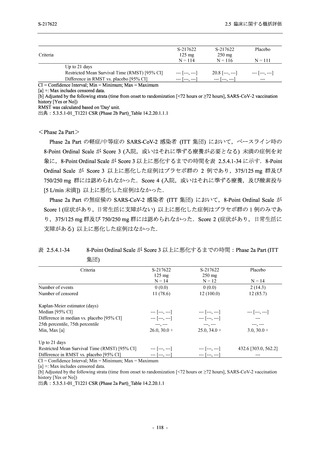
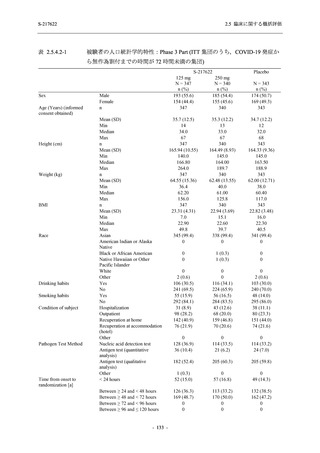
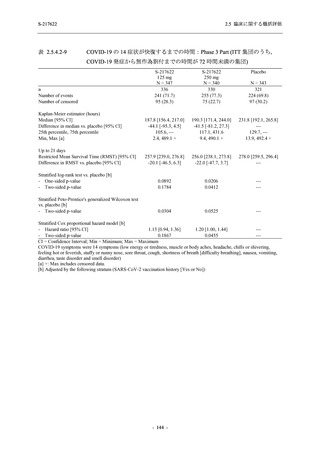
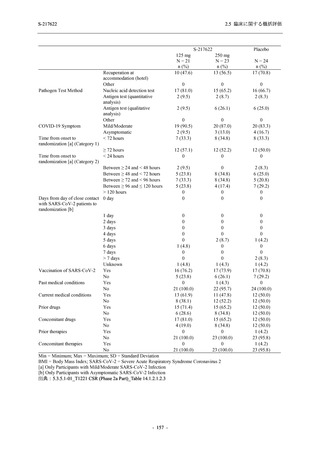
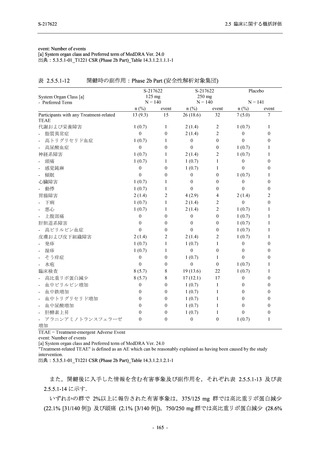
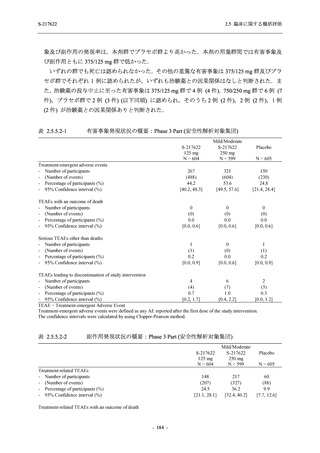
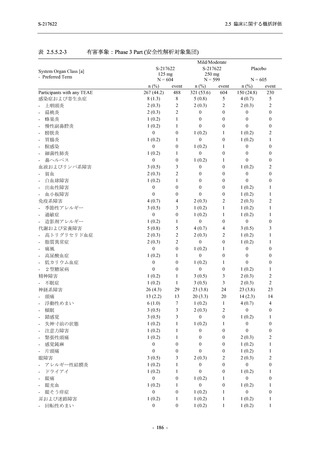
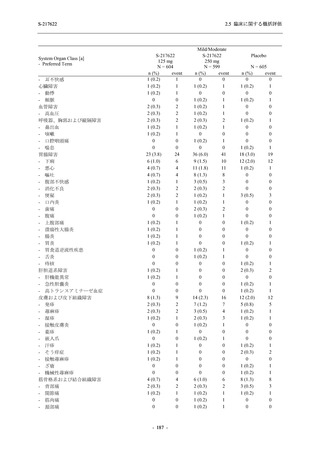
T1221 試験 Phase 3 Part において,死亡は認められず,その他の重篤な有害事象は 375/125 mg
群及びプラセボ群でそれぞれ 1 例に認められたが,いずれも治験薬との因果関係はなしと判断
された.有害事象の発現率は,375/125 mg 群 44.2% (267/604 例),750/250 mg 群 53.6% (321/599
例),プラセボ群 24.8% (150/605 例) (以下同順),副作用の発現率は 24.5% (148/604 例),36.2%
(217/599 例),9.9% (60/605 例) であり,有害事象及び副作用の発現率は,プラセボ群と比較して
本剤群で高く,本剤の用量群間では 375/125 mg 群の方が低かった.プラセボ群と比較して本剤
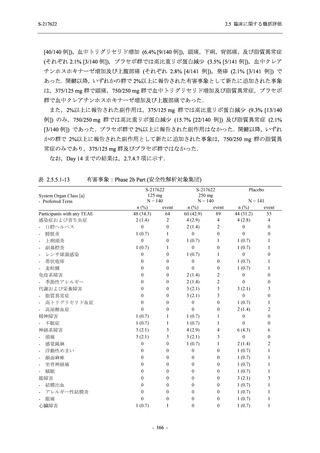
群で発現頻度の高い有害事象及び副作用の多くは臨床検査関連であり,Phase 2a Part 及び
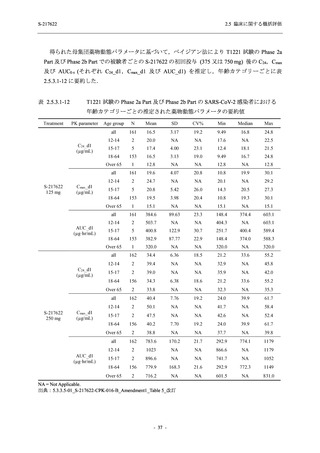
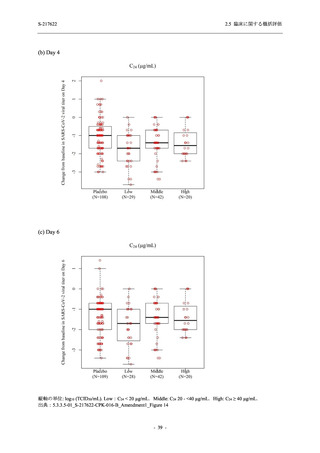
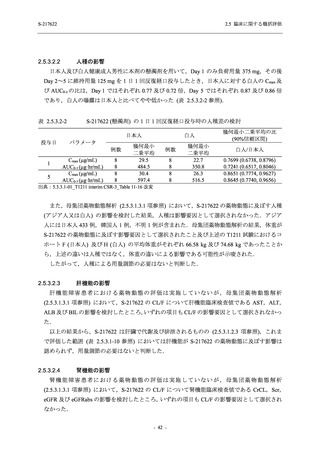
Phase 2b Part でも報告された事象であった.臨床検査値において,プラセボ群と比較して本剤群
で高比重リポ蛋白の減少,トリグリセリドの増加,総ビリルビン及び直接ビリルビンの増加,
血清鉄の増加,UIBC の低下が認められたが,いずれも一時的な変動であった.認められた有害
事象のほとんどは軽度であり.重症度が高度の事象では,750/250 mg 群の頭痛のみが因果関係
ありと判断されたがパラセタモール投与により回復し,治験薬の投与は継続された.治験薬の
投与中止に至った有害事象では,375/125 mg 群の湿疹及び嘔吐,750/250 mg 群の発疹 (2 件),
プラセボ群の筋力低下及び感覚鈍麻が治験薬との因果関係ありと判断されたが,いずれも治験
薬の投与中止後に軽快又は回復した.
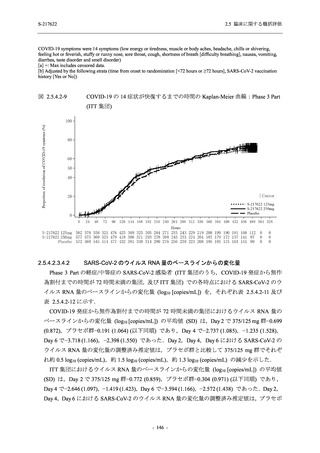
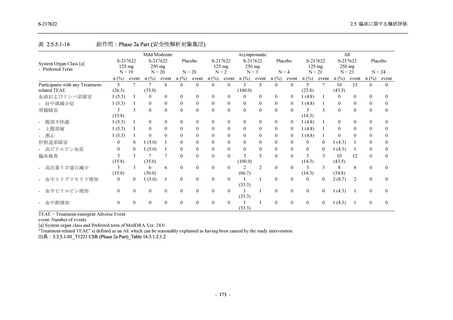
T1221 試験 Phase 2a Part,Phase 2b Part,及び Phase 3 Part を併合した集団でも,Phase 3 Part と
同様の傾向であった.
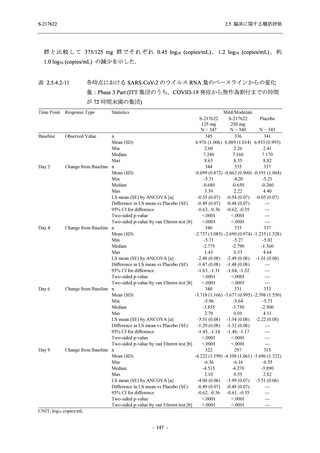
以上の結果から,T1221 試験 Phase 3 Part で新たに認められた安全性の懸念はなく,併合集団
でも同様の傾向であったことから,Phase 2a Part 及び Phase 2b Part の結果に基づく評価と同様
に,SARS-CoV-2 感染者における本剤の 1 日 1 回 5 日間の経口投与 (1 日目は 375 mg,2 日目か
ら 5 日目は 125 mg) での安全性に特段の懸念は認められず,忍容性は良好であった.
- 196 -
2.5.5.2.8
2.5 臨床に関する概括評価
安全性評価のまとめ
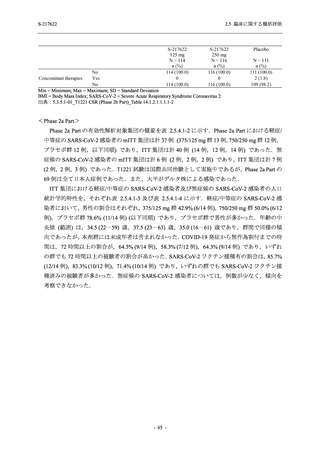
T1221 試験 Phase 3 Part において,死亡は認められず,その他の重篤な有害事象は 375/125 mg
群及びプラセボ群でそれぞれ 1 例に認められたが,いずれも治験薬との因果関係はなしと判断
された.有害事象の発現率は,375/125 mg 群 44.2% (267/604 例),750/250 mg 群 53.6% (321/599
例),プラセボ群 24.8% (150/605 例) (以下同順),副作用の発現率は 24.5% (148/604 例),36.2%
(217/599 例),9.9% (60/605 例) であり,有害事象及び副作用の発現率は,プラセボ群と比較して
本剤群で高く,本剤の用量群間では 375/125 mg 群の方が低かった.プラセボ群と比較して本剤
群で発現頻度の高い有害事象及び副作用の多くは臨床検査関連であり,Phase 2a Part 及び
Phase 2b Part でも報告された事象であった.臨床検査値において,プラセボ群と比較して本剤群
で高比重リポ蛋白の減少,トリグリセリドの増加,総ビリルビン及び直接ビリルビンの増加,
血清鉄の増加,UIBC の低下が認められたが,いずれも一時的な変動であった.認められた有害
事象のほとんどは軽度であり.重症度が高度の事象では,750/250 mg 群の頭痛のみが因果関係
ありと判断されたがパラセタモール投与により回復し,治験薬の投与は継続された.治験薬の
投与中止に至った有害事象では,375/125 mg 群の湿疹及び嘔吐,750/250 mg 群の発疹 (2 件),
プラセボ群の筋力低下及び感覚鈍麻が治験薬との因果関係ありと判断されたが,いずれも治験
薬の投与中止後に軽快又は回復した.
T1221 試験 Phase 2a Part,Phase 2b Part,及び Phase 3 Part を併合した集団でも,Phase 3 Part と
同様の傾向であった.
以上の結果から,T1221 試験 Phase 3 Part で新たに認められた安全性の懸念はなく,併合集団
でも同様の傾向であったことから,Phase 2a Part 及び Phase 2b Part の結果に基づく評価と同様
に,SARS-CoV-2 感染者における本剤の 1 日 1 回 5 日間の経口投与 (1 日目は 375 mg,2 日目か
ら 5 日目は 125 mg) での安全性に特段の懸念は認められず,忍容性は良好であった.
- 196 -