














































































































































よむ、つかう、まなぶ。
【資料No.1】2.5_臨床に関する概括資料 (203 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_29325.html |
| 出典情報 | 薬事・食品衛生審議会 薬事分科会(令和4年度第5回 11/22)、医薬品第二部会(令和4年度第13回 11/22)(合同開催)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
S-217622
2.5 臨床に関する概括評価
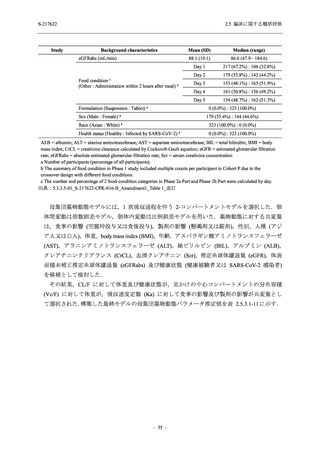
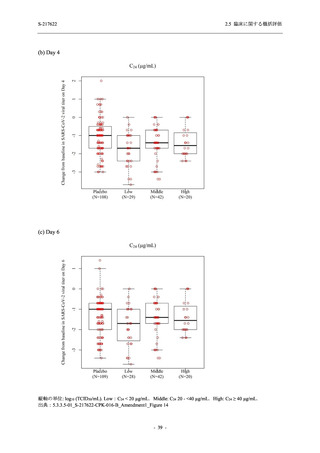
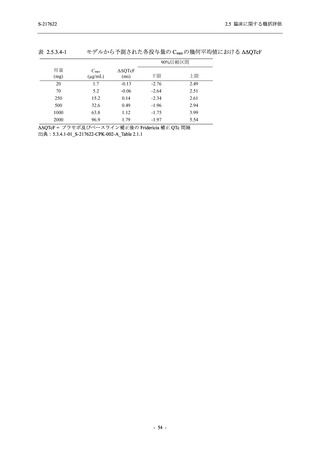
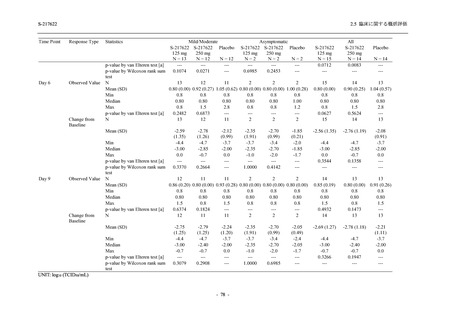
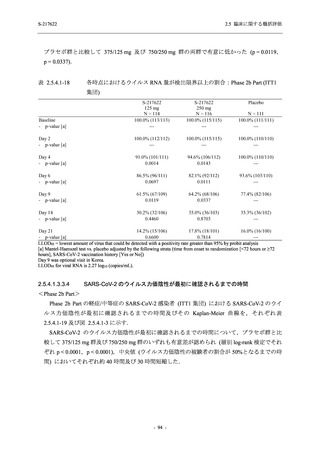
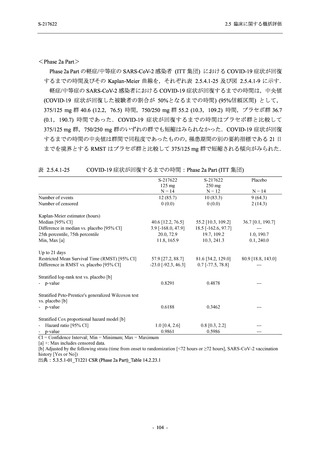
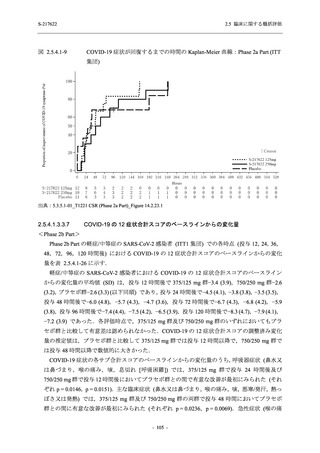
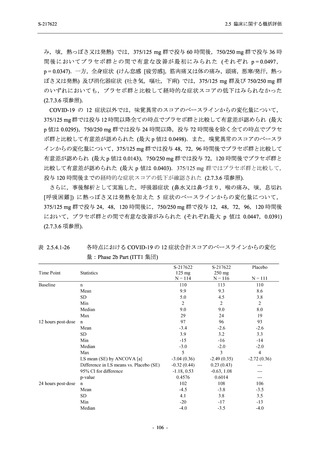
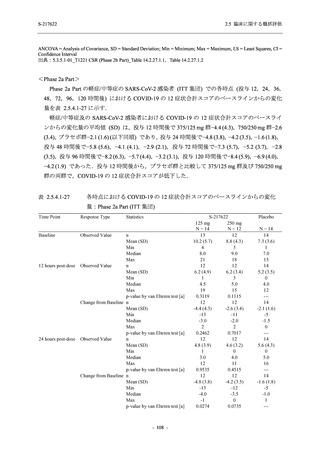
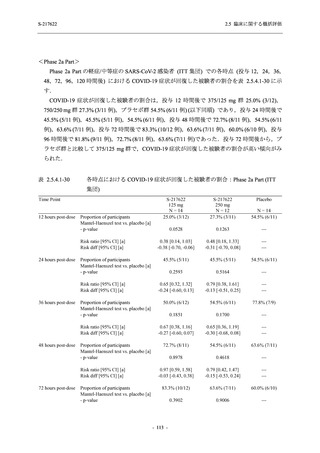
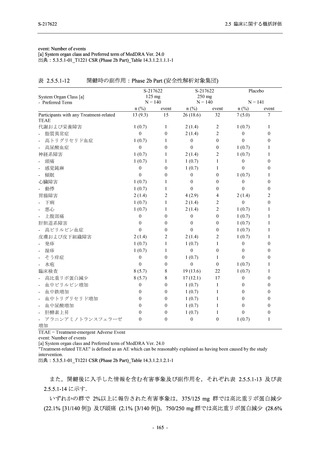
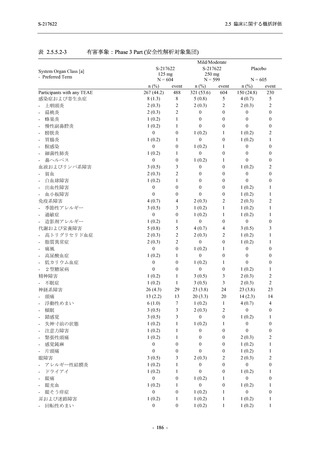
断された.また,開鍵後に入手した情報を含めると,高比重リポ蛋白減少 (22.1% [31/140 例])
及び頭痛 (2.1% [3/140 例]) であり,それぞれ 9.3% (13/140 例) 及び 0.7% (1/140 例) が因果関係
ありと判断された.
Phase 2a Part において,死亡を含む重篤な有害事象は認められていない (2.5.5.1.2.2 項参照).
申請用法用量 (375/125 mg 群) で 3 例以上に報告された有害事象は,高比重リポ蛋白減少
(14.3% [3/21 例]) のみであった.いずれも因果関係ありと判断されたが,重症度は軽度であり,
医学的処置なく回復した (2.5.5.1.2.1 項及び 2.7.4.7 項参照).
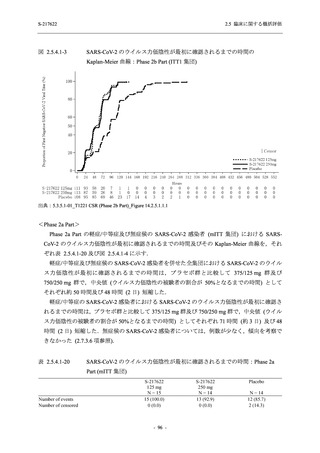
健康成人を対象とした T1211 試験及び T1215 試験においても,死亡を含む重篤な有害事象及
び高度な有害事象は認められておらず,申請用法用量において,本剤は概ね安全かつ忍容性は
良好であった (2.5.5.1.5 項参照).
以上の結果から,患者における情報は限定的であるものの,現時点では,申請用法用量での
本剤の安全性に特段の懸念は認められなかった.本剤の市販後には,一般使用成績調査を行い,
使用実態下での患者の安全性を確認することを計画している.
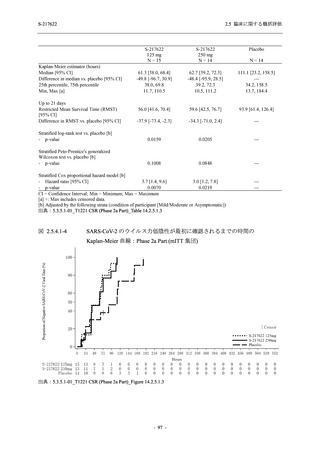
T1221 試験 Phase 3 Part の結果に基づき,再検討した内容を追記する.
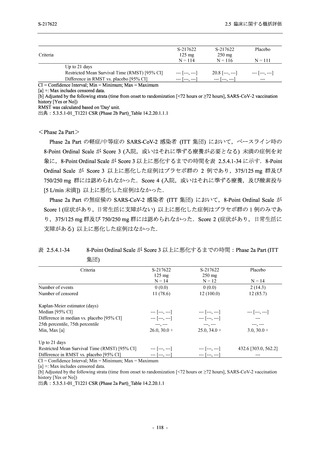
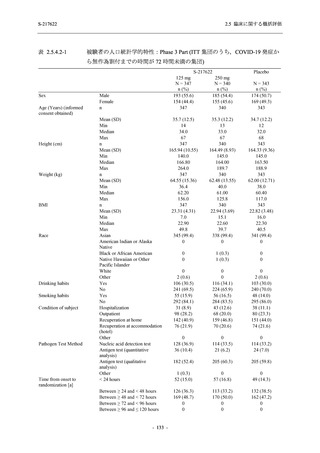
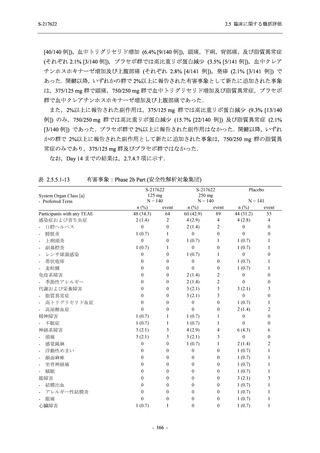
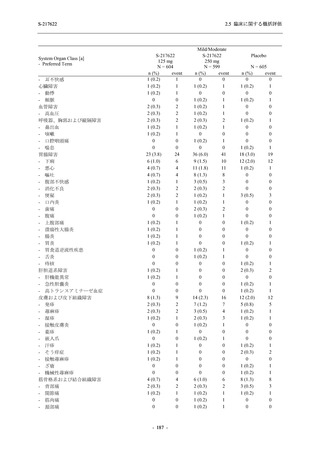
T1221 試験 Phase 3 Part において,死亡は認められず,その他の重篤な有害事象は 375/125 mg
群及びプラセボ群でそれぞれ 1 例に認められたが,いずれも治験薬との因果関係はなしと判断
された.
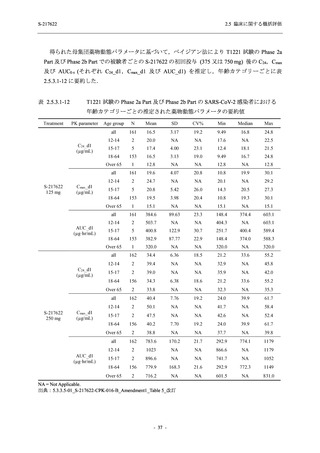
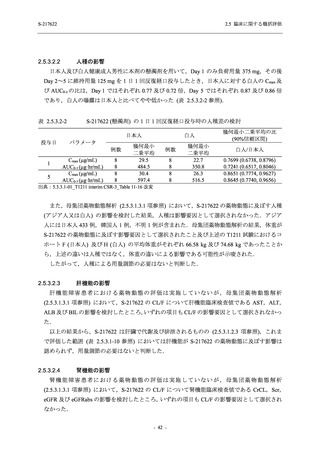
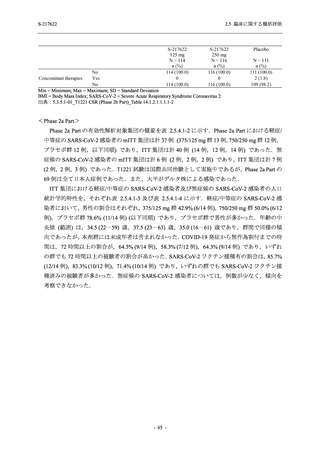
申請用法用量 (375/125 mg 群) での有害事象の発現率は,44.2% (267/604 例) であった.
発現頻度の高い有害事象及び副作用の多くは臨床検査関連であり,臨床検査値を評価した結果,
プラセボ群と比較して本剤群で高比重リポ蛋白の減少,トリグリセリドの増加,総ビリルビン
及び直接ビリルビンの増加,血清鉄の増加,UIBC の低下が認められたが,いずれも一時的な変
動であった.また,報告された有害事象のほとんどは軽度又は中等度であった.重症度が高度
の事象は 375/125 mg 群で重度月経出血が 1 例に認められたが,治験薬との因果関係はなしと判
断された.
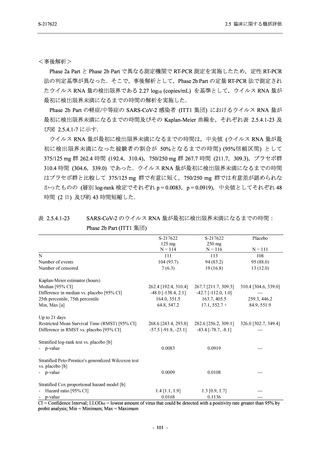
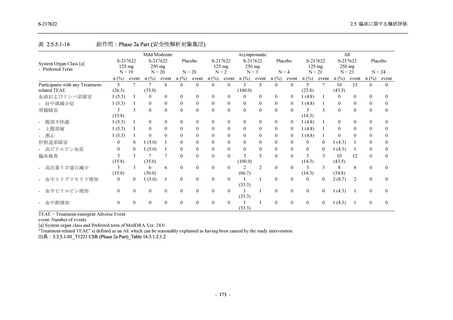
Phase 2a Part,Phase 2b Part,及び Phase 3 Part を併合した申請用法用量 (375/125 mg 群) での
有害事象の発現率は 42.6% (325/763 例) であり,本剤の安全性は Phase 3 Part と同様の傾向で
あった.
以上の結果から,T1221 試験 Phase 3 Part では,Phase 2a Part 及び Phase 2bPart の結果に基づく
評価から変更なく,申請用法用量での本剤の安全性に特段の懸念は認められなかった.本剤の
市販後には,一般使用成績調査を行い,使用実態下での患者の安全性を確認することを計画し
ている.
2.5.6.4
ベネフィット・リスク評価
本資料は,T1211 試験コホート A からコホート S まで,T1215 試験,T1221 試験 Phase 2b Part
及び Phase 2a Part の結果に基づいて記載した.申請用法用量での本剤の抗ウイルス効果及び臨
床症状改善効果が確認され,安全性に特段の懸念は認められていないことから,本剤は 1 日 1
回 5 日間の経口投与により,SARS-CoV-2 による感染症に対する有効な治療の選択肢となり得
ると考える.
- 203 -
2.5 臨床に関する概括評価
断された.また,開鍵後に入手した情報を含めると,高比重リポ蛋白減少 (22.1% [31/140 例])
及び頭痛 (2.1% [3/140 例]) であり,それぞれ 9.3% (13/140 例) 及び 0.7% (1/140 例) が因果関係
ありと判断された.
Phase 2a Part において,死亡を含む重篤な有害事象は認められていない (2.5.5.1.2.2 項参照).
申請用法用量 (375/125 mg 群) で 3 例以上に報告された有害事象は,高比重リポ蛋白減少
(14.3% [3/21 例]) のみであった.いずれも因果関係ありと判断されたが,重症度は軽度であり,
医学的処置なく回復した (2.5.5.1.2.1 項及び 2.7.4.7 項参照).
健康成人を対象とした T1211 試験及び T1215 試験においても,死亡を含む重篤な有害事象及
び高度な有害事象は認められておらず,申請用法用量において,本剤は概ね安全かつ忍容性は
良好であった (2.5.5.1.5 項参照).
以上の結果から,患者における情報は限定的であるものの,現時点では,申請用法用量での
本剤の安全性に特段の懸念は認められなかった.本剤の市販後には,一般使用成績調査を行い,
使用実態下での患者の安全性を確認することを計画している.
T1221 試験 Phase 3 Part の結果に基づき,再検討した内容を追記する.
T1221 試験 Phase 3 Part において,死亡は認められず,その他の重篤な有害事象は 375/125 mg
群及びプラセボ群でそれぞれ 1 例に認められたが,いずれも治験薬との因果関係はなしと判断
された.
申請用法用量 (375/125 mg 群) での有害事象の発現率は,44.2% (267/604 例) であった.
発現頻度の高い有害事象及び副作用の多くは臨床検査関連であり,臨床検査値を評価した結果,
プラセボ群と比較して本剤群で高比重リポ蛋白の減少,トリグリセリドの増加,総ビリルビン
及び直接ビリルビンの増加,血清鉄の増加,UIBC の低下が認められたが,いずれも一時的な変
動であった.また,報告された有害事象のほとんどは軽度又は中等度であった.重症度が高度
の事象は 375/125 mg 群で重度月経出血が 1 例に認められたが,治験薬との因果関係はなしと判
断された.
Phase 2a Part,Phase 2b Part,及び Phase 3 Part を併合した申請用法用量 (375/125 mg 群) での
有害事象の発現率は 42.6% (325/763 例) であり,本剤の安全性は Phase 3 Part と同様の傾向で
あった.
以上の結果から,T1221 試験 Phase 3 Part では,Phase 2a Part 及び Phase 2bPart の結果に基づく
評価から変更なく,申請用法用量での本剤の安全性に特段の懸念は認められなかった.本剤の
市販後には,一般使用成績調査を行い,使用実態下での患者の安全性を確認することを計画し
ている.
2.5.6.4
ベネフィット・リスク評価
本資料は,T1211 試験コホート A からコホート S まで,T1215 試験,T1221 試験 Phase 2b Part
及び Phase 2a Part の結果に基づいて記載した.申請用法用量での本剤の抗ウイルス効果及び臨
床症状改善効果が確認され,安全性に特段の懸念は認められていないことから,本剤は 1 日 1
回 5 日間の経口投与により,SARS-CoV-2 による感染症に対する有効な治療の選択肢となり得
ると考える.
- 203 -